„Hat SARS-CoV-2 den Gipfel seines Fitnessbergs erreicht? – Wir wissen es nicht.“
Im Corona-Gespräch: Emma Hodcroft, Bern
Gespräch Henrik Müller (8. Februar 2021)
(08.03.2021) Emma Hodcrofts Expertise liegt in der Phylogenetik und molekularen Epidemiologie viraler Pathogene. Am Institut für Sozial- und Präventivmedizin der Universität Bern arbeitet sie als Teil des NextStrain-Teams an SARS-CoV-2. Im Gespräch ordnet sie den Beitrag menschlichen Verhaltens zur Virus-Evolution ein, erklärt, warum sich die Welt auf britische Sequenzierdaten verlässt, und verdeutlicht, was sich in der Pandemiebekämpfung ändern muss.
Laborjournal: Wie alle RNA-Viren entwickelt SARS-CoV-2 schnell sogenannte Fluchtmutanten. Wie viele bedeutsame Varianten kursieren derzeit?
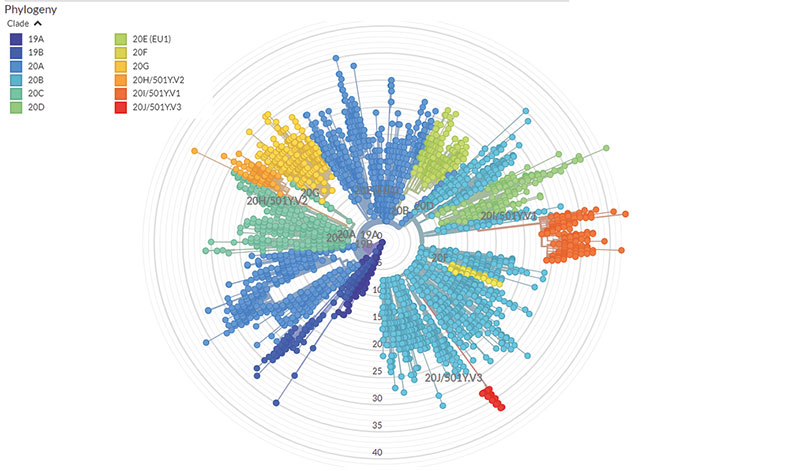
Emma Hodcroft » Momentan erregen drei Varianten unsere Besorgnis: 20I/501Y.V1, 20H/501Y.V2 und 20J/501Y.V3 – oder wie von den Medien genannt: die „britische“ Variante B.1.1.7, die „südafrikanische“ Variante B.1.351 und die „brasilianische“ Variante P.1. Laut epidemiologischer Modellierungen ist B.1.1.7 tatsächlich um vierzig bis siebzig Prozent leichter übertragbar, B.1.351 dagegen um fünfzig bis sechzig Prozent. Allerdings wissen wir wenig darüber, welche ihrer Mutationen uns wie genau auf pandemischer und klinischer Ebene beeinflussen.
Was unterscheidet diese Varianten genotypisch von 20A.EU1 – also der Virusvariante, die letzten Sommer in Europa vorherrschte?
Hodcroft » 20A.EU1 entstand in Spanien und breitete sich rasch über Europa aus. Noch immer ist sie hier die dominante Variante. Übertragbarer als die bis dahin dominierenden Varianten ist sie allerdings nicht. Vielmehr können wir die Ausbreitung mit dem menschlichen Reiseverhalten und der Lockerung von Beschränkungen erklären. Die Variante B.1.1.7 entwickelte sich dagegen Ende letzten Jahres im Südosten Englands – und dort stiegen die Infektionszahlen trotz des November-Lockdowns. Es verbreitet sich nicht nur infolge menschlicher Verhaltensmuster, sondern auch infolge der eigenen viralen Eigenschaften. SARS-CoV-2 erwirbt im Durchschnitt zwei Mutationen pro Genom pro Monat. B.1.1.7 jedoch beherbergt mehr Mutationen...
..., welche die virale Fitness auf biochemischer Ebene verbessern.
Hodcroft » Genau. Die meisten Mutationen finden sich im Spike-Protein, was natürlich Sinn macht, da dessen Rezeptor-bindende Domäne mit ACE2-Rezeptoren des Wirts interagiert, um die entsprechenden Zellen zu infizieren. Warum bestimmte Mutationen funktionell relevant sind, wissen wir aber meist nicht, sondern wir stolpern immer wieder über sie. Die Häufigkeiten der Spike-Mutationen N501Y, N501T und N501S waren ja sogar namensgebend für die drei 501-Varianten.
Entstammen sie folglich derselben Klade?
Hodcroft » Nein, sie entwickelten sich aus unterschiedlichen Kladen – konkret aus denjenigen mit den Bezeichnungen 20B und 20C. Und sie unterscheiden sich maßgeblich, vor allem in einer Mutation an der Position E484 des Spike-Proteins, die sich nicht in der „britischen“ B.1.1.7, sondern nur in der „südafrikanischen“ B.1.351 und der „brasilianischen“ P.1 finden lässt. Besorgnis erregt diese Mutation, da beide Varianten laut Laborbefunden Menschen effektiver reinfizieren.
Sind auch bedeutsame Mutationen außerhalb des Spike-Proteins bekannt?
Hodcroft » Tatsächlich erwirbt das Virus gegenwärtig eine zunehmende Anzahl polymorpher Nukleotid-Positionen in verschiedenen Leserastern seines Genoms – neben Spike vor allem im Nukleokapsid-Protein, aber auch in Nicht-Oberflächen-Proteinen. Mutationen im Open Reading Frame 8 (ORF8) zum Beispiel verstümmeln dessen Proteinprodukt. Vorläufige Studien deuten auf einen schwereren klinischen Verlauf dieser Variante hin. Für die meisten Mutationen kennen wir deren Auswirkungen aber nicht. Im Moment scheinen jedenfalls Mutationen in Spike ausschlaggebend.
Welche Art von Mutation würde SARS-CoV-2 ungleich gefährlicher machen?
Hodcroft » Mutationen treten selten vereinzelt auf. Entsprechend schwierig ist es, sie einzeln für phänotypische Ausprägungen verantwortlich zu machen. Allerdings haben wir Hinweise auf mehrere mögliche Mechanismen. Eine der bekanntesten Mutationen in Spike ist D641G, die bereits im Januar 2020 entstand und sich in 98 Prozent aller Viren findet. Sie hält Spike länger in einer offenen Konformation, wodurch das Virus öfter an ACE2 binden kann. Andere Mutationen verschleiern virale Proteine wiederum vor Immunkomponenten, sodass das Virus der Körperabwehr länger ausweichen oder Menschen reinfizieren kann.
Die RNA-abhängige RNA-Polymerase von SARS-CoV-2 verfügt über einen Fehlerkorrekturmechanismus. Allerdings korrigiert dieser nur Substitutionen aber keine Deletionen, wie etwa Spikes Δ69-70 in B.1.1.7 oder Δ242-244 in B.1.351. Verbessern Deletionen also am ehesten die virale Fitness?
Hodcroft » Das ist unwahrscheinlich, denn der virale Korrekturlesemechanismus mag vielleicht keine Deletionen korrigieren, die natürliche Selektion wirkt aber gleichermaßen auf alle Mutationsarten. Unabhängig davon ist Δ69-70 tatsächlich sehr erfolgreich. Diese Deletion verkürzt eine Oberflächenschleife auf Spikes Außenseite und macht sie schwerer für Antikörper zugänglich.
Wie sehr beeinflussen Mutationen die Basisreproduktionszahl von SARS-CoV-2?
Hodcroft » Auf molekularer Ebene wissen wir zu wenig, um das zu sagen. Zwar können wir bestimmte Virus-Varianten mit Inzidenzraten korrelieren, aber nicht auf bestimmte Mutationen eingrenzen.
Es ist also unbekannt, welche molekularen Mechanismen bestimmte Varianten übertragbarer machen, Inkubationszeiten verkürzen oder zu Reinfektionen führen?
Hodcroft » Laut britischen Untersuchungen erhöht 501Y.V1 die Viruslast. Jeder Atemzug setzt dann mehr Viren frei, die wiederum mehr Menschen infizieren können. Daten zu Inkubationszeiten zeigen dagegen zu hohe Schwankungsbreiten, um eine Aussage zu treffen. Wir wissen einfach zu selten, wann genau sich jemand infiziert oder das Virus verbreitet. Die Frage nach den Reinfektionen dagegen betrifft aktuell vor allem die Varianten, die ausgehend von Südafrika und Brasilien zirkulieren. Blutplasma rekonvaleszenter Personen neutralisiert 501Y.V2 und 501Y.V3 zehnmal schlechter, was Reinfektionen wahrscheinlicher macht. Ob wir diese Laborbefunde auf den gesamten menschlichen Organismus extrapolieren können, bleibt aber abzuwarten. Reinfektionen sind ja vereinzelt auch von älteren Varianten bekannt.
Woher beziehen Sie überhaupt die Sequenzdaten aller Varianten?
Hodcroft » Von GISAID, also der Global Initiative on Sharing All Influenza Data, mit Sitz in München. Auf deren Online-Plattform stellen Labore ihre Sequenzierungsdaten kostenlos zur Verfügung. Was erstaunlich gut funktioniert, wenn man bedenkt, dass unser wissenschaftliches Anreizsystem nicht gerade dazu ermuntert, unveröffentlichte Daten frei nutzbar zu machen.
Unser Wissen über neue Varianten hängt demnach von der Gutmütigkeit der Sequenzierlabore ab?
Hodcroft » Komplett! Woraus wir jetzt lernen müssen, dass Sequenzierleistungen nicht nur durch europäische Förderagenturen, sondern auch das öffentlich-rechtliche Gesundheitswesen finanziert werden sollten. Geldgeber wie die US-amerikanischen National Institutes of Health (NIH) und das Center of Disease Control and Prevention (CDC) verpflichten beispielsweise bereits dazu, Sequenzen frei verfügbar zu machen. Schließlich sind solche Daten in einer Pandemie im wahrsten Sinn des Wortes überlebenswichtig. Europa sollte für potenzielle zukünftige Pandemien vielleicht nicht nur auf Gutmütigkeit vertrauen.
Sie selbst arbeiten aktuell Vollzeit an SARS-CoV-2 als Teil des sogenannten NextStrain-Teams. Auch NextStrain stellt seine Analysedaten frei zur Verfügung. Was steckt genau dahinter?
Hodcroft » NextStrain ist ein Open-Source-Projekt, mit dessen Bioinformatik-Tools sich die Evolution der Genomdaten bekannter Pathogene in Echtzeit verfolgen und visualisieren lässt, und zwar für Dengue-, Influenza-, Mumps-, Masern-, Westafrikanische Ebola-, West-Nil- und Zika-Viren sowie für Enterovirus D68, Mycobacterium tuberculosis – und natürlich für SARS-CoV-2.
NextStrain verfolgt also die Entstehung neuer Sequenzvarianten und ihre Übertragungsketten?
Hodcroft » Ganz genau. Wir vergleichen Mutationshäufigkeiten und Mutationsraten und schlussfolgern aus den zugehörigen Sequenzdistanzen Verwandtschaftsverhältnisse. Indem wir phylogenetische Stammbäume um Ort und Zeit der Probenentnahme erweitern, können wir auf Genom und Verbreitung von Vorläufervarianten extrapolieren. Damit lassen sich spannende Fragen zur geographischen und zeitlichen Evolution von Pathogenen beantworten: Warum finden virale Ausbrüche in unregelmäßigen Wellen statt? Warum unterscheidet sich die Influenza-Saison von Jahr zu Jahr? Warum dominieren bestimmte Sequenzvarianten plötzlich ein ganzes Land? Und brandaktuell: Wie effektiv begegnet das Immunsystem bestimmten Varianten? Wie weit können wir uns im phylogenetischen Baum entfernen, bis entsprechende Antikörper ein Virus oder eine Variante nicht mehr erkennen?
Bekannt ist NextStrain vielleicht am ehesten für seine interaktiven Abbildungen zu SARS-CoV-2-Varianten...
Hodcroft » Ja, unter nextstrain.org stehen Applikationen für eigene phylogenetische Analysen bereit. Statt einzelne Informationsbrocken mühsam in PubMed zusammensuchen zu müssen, stellt NextStrain Genomdaten inklusive ihrer Zeitauflösung, ihrer Länderinformation, ihrer Mutationen und Aminosäure-Austausche zentral an einem Ort in einer graphisch aufgearbeiteten Schnittstelle bereit.
Erlaubt es dieses deskriptive Level, Fitnessvorteile für SARS-CoV-2 zu modellieren, zum Beispiel auch mit Blick auf die Vakzin-Entwicklung?
Hodcroft » Tatsächlich war es die Gründungsidee von NextStrain, das ursprünglich NextFlu hieß, die Evolution des Influenza-Genoms zu verstehen, um als Teil des globalen Influenza-Programms der WHO bessere Grippeimpfstoffe zu ermöglichen. Für SARS-CoV-2 fehlen uns dafür aber noch repräsentative Proben jeden Landes. Deshalb schauen wir uns bisher vor allem globale Verzweigungs-Indices an, analysieren also, wie schnell bestimmte Äste des phylogenetischen Baumes wachsen. Denn Impfstoffe sollten wir natürlich auf evolutionär erfolgreiche Varianten ausrichten.
Auf wie viele SARS-CoV-2-Genome können sie bisher zurückgreifen?
Hodcroft » Mittlerweile stehen uns eine halbe Million Sequenzen von SARS-CoV-2 zur Verfügung, was erstaunlich für dessen Alter ist. Um das in die richtige Perspektive zu rücken: Vor der Pandemie habe ich an Enterovirus D68 gearbeitet. Es ist seit 1962 bekannt, doch bisher haben wir gerade mal achthundert Genomsequenzen. So unglaublich unsere Datenmenge für SARS-CoV-2 ist, wirft sie jedoch auch Probleme auf. Keiner unserer Algorithmen und Analyseprozesse ist darauf ausgelegt, eine halbe Million Sequenzen in Echtzeit, also am besten jeden Tag, zu analysieren. Für tägliche Situations-Updates müssen wir uns auf Stichproben von fünf- bis sechstausend Sequenzen beschränken.
Was macht eine Parallelisierung so schwierig?
Hodcroft » Nichts. Projekte zur Entwicklung schnellerer Algorithmen wurden bisher halt nicht gefördert. Erst die Pandemiesituation bringt deren Wichtigkeit in den Fokus. Über-Nacht-Algorithmen für Millionen Sequenzen werden womöglich den Kampf gegen zukünftige Krankheitserreger entscheiden.
Wie sehr hängen NextStrains Aussagen dann von der gewählten Stichprobe ab?
Hodcroft » Nähmen wir zufällige Stichproben, würden globale Aussagen von denjenigen Ländern dominiert, die mehr Sequenzen als andere Länder beisteuern. Dann sähe es so aus, als stammten alle Coronavirus-Probleme der Welt aus Großbritannien, da von dort die Hälfte aller bisherigen Sequenzen stammt. Die Briten sequenzieren unglaublich viele Genome. Aus diesem Grund gewichten wir Stichproben relativ zur Rolle ihres Ursprungsorts. Etwa indem wir die gleiche Anzahl an Sequenzen für verschiedene Länder und unterschiedliche Zeitpunkte verwenden, unabhängig davon wie viele Proben jeweils tatsächlich verfügbar wären. Mit nur einigen tausend von fünfhunderttausend Sequenzen übersehen wir damit natürlich einiges auf globaler Ebene. Deshalb führen wir auch für jeden Kontinent und jedes Land regionale Analysen durch, falls von dort genug Sequenzen vorhanden sind. Für Länder ohne Sequenzproben können wir natürlich keine Aussagen treffen.
Wieso stammen die meisten Genomsequenzen aus Großbritannien?
Hodcroft » Weil das britische Gesundheitssystem die Wichtigkeit landesweiter Genomsequenzierungen bereits im Frühjahr 2020 erkannte. Dortige Forschungslabore und Kliniken errichteten binnen zwei Monaten in einer landesweiten Anstrengung ein Netzwerk von Sequenzierzentren. Neben Impfstoffen war das sozusagen das britische „Moonshot“-Programm. Es ist umwerfend, was Großbritannien erreicht hat. Und jetzt zahlt es sich aus, denn die Briten haben den besten Überblick über die Pandemie. Und der Rest von uns verlässt sich auf ihre Sequenzierdaten, um neue Varianten zu identifizieren.
Warum gerade Großbritannien? Der Rest der Welt verfügte letztes Frühjahr doch über den gleichen Wissensstand...
Hodcroft » Andere Länder scheuten aber eine so riesige Investition, weil sie glaubten, die Pandemie sei in ein paar Monaten vorbei. Zwar gab es dort auch Wissenschaftler, die den Nutzen von Sequenzierungszentren frühzeitig verstanden, aber sie trafen in ihren Regierungen nicht auf die Bereitschaft, einen Haufen Geld auf deren Expertise zu wetten.
Wie hoch ist der britische Sequenzier-Durchsatz?
Hodcroft » Je nach Anzahl Corona-positiver Proben sind es zwischen zwei und zehn Prozent. Natürlich hat auch Großbritannien eine harte Grenze verfügbarer Sequenzierautomaten. Bisher haben sie dennoch eine Viertelmillion Genomsequenzen beigesteuert.
Wären Punktmutations-Assays vielleicht eine Alternative zu den kostenintensiven Genomsequenzierungen?
Hodcroft » Für die schnelle Suche nach bekannten Varianten sind sie sicher besser geeignet. Allerdings finden sie keine neuen Mutationen, weshalb sie für die Überwachung von Sequenzvarianten und das Verstehen von genetischen Mustern keinen Ersatz darstellen. Genomsequenzierungen sind hier die einzige Möglichkeit.
Wie wird also das Genom von SARS-CoV-2 in Zukunft aussehen?
Hodcroft » Das ist die Millionen-Dollar-Frage. Bisher haben wir gelernt, dass das Virus übertragbarer werden, dem Immunsystem ausweichen, Menschen reinfizieren und Impfstoffe umgehen kann. Die große Frage ist jetzt: Hat SARS-CoV-2 den Gipfel seines Fitnessbergs erreicht? Wir wissen es nicht.
Welche weiteren Faktoren entschleunigen die Evolution von SARS-CoV-2?
Hodcroft » Seine Mutationsrate ändert sich nicht. Beeinflussen können wir nur, welche Mutationen vorherrschen. Das hängt offensichtlich mit der natürlichen Selektion vorteilhafter Mutationen zusammen. Deshalb dürfen wir dem Virus nur möglichst wenig Spielraum bieten, seine eigenen Möglichkeiten zu erforschen und zu mutieren. Dafür sind niedrige Inzidenzraten entscheidend. Nur so verwehren wir ihm Umgebungen, aus denen es profitieren könnte – wie etwa immungeschwächte oder medikamentös behandelte Menschen. Gleichzeitig müssen wir intensiv nach neuen Variationen Ausschau halten. Nur dann können wir schnell reagieren. Denn leider korrelieren Orte mit schlechten Sequenzier-Kapazitäten und solche mit großen Ausbrüchen in der Vergangenheit.
Lassen sich intrinsisch übertragbarere Varianten überhaupt von epidemiologischen Faktoren wie etwa Superspreading Events abgrenzen, die ja ebenfalls für Anstiege der Fallzahlen sorgen?
Hodcroft » Das ist schwierig. Und ist ein Grund, warum manche Schlagzeile zur Übertragbarkeit neuer Varianten in den letzten Wochen unnötig für Aufregung gesorgt hat. Denn allzu selten erzählen sie die ganze Geschichte. Der wichtigste Faktor ist nämlich tatsächlich nach wie vor das menschliche Verhalten. Europas dominierende Variante 20A.EU1 beispielsweise ist im Labor nicht übertragbarer als andere Varianten. Erst das Reiseverhalten der Menschen und die Lockerungen der Kontaktbeschränkungen erlaubten seine Verbreitung. Nur was übrig bleibt, wenn Modellierungsanalysen den Faktor Mensch ausschließen, liegt alleine am Virus selbst.
Was bedeutet das konkret für B.1.1.7, B.1.351 und P.1?
Hodcroft » Laut britischen Modellrechnungen können wir die Ausbreitung von B.1.1.7 nicht komplett durch menschliches Verhalten erklären. Für die anderen sind noch keine endgültigen Schlussfolgerungen möglich. Wie erwähnt legen vorläufige Laborversuche nahe, dass ihre einzigartigen Mutationen tatsächlich die Funktionsweise des Virus beeinflussen. Trotzdem sollten die Medien mit den Aussagen ihrer schnellen Schlagzeilen vorsichtiger sein.
Könnte ein Selektionsdruck durch Impfstoffe diese Varianten schneller evolvieren lassen?
Hodcroft » Dafür sollten wir erstmal die zwei Extreme betrachten. Ist niemand geimpft, unterliegt das Virus keinem impfbedingten Selektionsdruck. Sind alle geimpft, kann es nicht zirkulieren und somit auch nicht evolvieren. Gefährlich ist das Zwischenszenario einer teilweise geimpften Bevölkerung. Diese Phase müssen wir schnell und unter geringen Inzidenzraten durchschreiten. Denn nur wenn SARS-CoV-2 zirkuliert, kann es sich anpassen. Die meisten Länder wollen den Großteil ihrer Bevölkerung vor dem Herbst impfen. Das ist, denke ich, die richtige Geschwindigkeit. Wobei sich Herstellungs- und Verteilungsprobleme in solch einem logistischen Albtraum von Milliarden notwendigen Impfdosen wahrscheinlich nicht vermeiden lassen. Umso wichtiger ist es, Impfpläne zu kommunizieren und auf Engpässe aufmerksam zu machen.
Werden wir am Ende in einem Influenza-ähnlichen Szenario jährlicher Impfwiederholungen landen?
Hodcroft » Auch das hängt von der derzeitigen Fitness des Virus ab. Hat es seinen Höhepunkt bereits erreicht, müssen wir Impfstoffe vielleicht erst in Jahren aktualisieren. Kann es weiter evolvieren, müssen wir ein Grippe-Szenario durchspielen. Der wichtigste Aspekt ist aber erneut nicht das Virus: Die Bevölkerung Europas können wir vielleicht bis Ende des Jahres impfen. Was aber ist mit den finanzschwachen Ländern im Rest der Welt? Solange SARS-CoV-2 dort zirkuliert, sind wir alle verwundbar. Denn entwickeln sich dort potenziell Impfstoff-immune Stämme, bedrohen sie uns alle. Deshalb hinkt der Vergleich mit Influenza. Jeder hatte schon mal eine Grippe, wir alle sind gleich immun. Für endemische saisonale Beta-Coronaviren gilt das zwar auch, für SARS-CoV-2 sind aber die meisten Menschen anfällig. Ließen wir es durch die Bevölkerung wüten, käme es irgendwann zu einem Grippe-Szenario – allerdings erst nach Millionen Toten. Die Frage ist: Wie erreichen wir es ohne Tote und ohne Impfstoff-immune Stämme? Nur, indem wir die Vakzinen gerechter und gleichmäßiger verteilen.
Hätten wir bisher etwas anders machen sollen?
Hodcroft » Europa wurde im letzten Sommer selbstgefällig. Das ist zwar verständlich, weil wir alle die Nase von der Pandemie voll hatten, aber seit Herbst bezahlen wir dafür. Wir haben Verhaltensänderungen infolge des Jahreszeitenwechsels nicht ernst genommen. Wir haben neue Varianten gesät. Wir haben exponentielles Wachstum unterschätzt. Länder in Asien und Ozeanien zeigen uns, wie es besser funktioniert. Die Leute dort führen im Allgemeinen ein normales Leben. Im Fall eines Ausbruchs riegeln sie sofort alles drakonisch für zwei, drei Wochen ab. Dann setzen sie ihr normales Leben fort. Warum sollte das nicht auch Europa bewerkstelligen können? Unsere halbgaren Sperrmaßnahmen dagegen haben kein Verfallsdatum. Wir haben keine Ahnung, wie lange wir die Fallzahlen im Auge behalten müssen. Und dieses Nichtwissen, dieser endlose Lockdown ist mental schwer zu ertragen und wirtschaftlich von den Unternehmen unmöglich zu tolerieren. Lokale Lockdowns mit Verfallsdaten, um einzelne Fälle unter Kontrolle zu bringen, wären für die Bevölkerung einfacher zu akzeptieren. Dann gäbe es nicht viel, wogegen jemand protestieren könnte. Viel wichtiger aber: Wir kämen der Ausrottung des Virus einen Riesenschritt näher.