Abgezocktes Nukleotid-Quartett
Nanoporensequenzierung
Thorsten Lieke
Die Sequenzierung von DNA mit Nanoporen ist noch zu ungenau. Ein Kartenspieler-Trick erhöht die Präzision.
Die Sequenzierung des menschlichen Genoms durch das im April 2003 abgeschlossene Humangenom-Projekt, kostete noch mehr als drei Milliarden Dollar. Schon ein Jahr später rief das National Human Genome Research Institute (NHGRI) in den USA das Ziel aus, die Kosten innerhalb von zehn Jahren auf unter 1.000 Dollar zu drücken.
Zum damaligen Zeitpunkt war dies eine sehr ambitionierte Vorgabe. Die Arbeiten an alternativen Sequenziermethoden liefen jedoch schon während des Humangenom-Projekts auf Hochtouren und mündeten letztlich in den heute gebräuchlichen Verfahren der Next-Generation-Sequenzierung. Aber auch die modernsten Sequenzierer können nicht ganz auf Polymerasen und Fluoreszenz-Farbstoffe verzichten. Bei mehr als sechs Milliarden Basenpaaren im diploiden Säugetiergenom, stellen diese aber noch immer einen erheblichen Kostenfaktor dar.
Billig und schnell...
Nicht zuletzt aus Kostengründen rückt deshalb die Nanoporensequenzierung immer mehr in den Blickpunkt, deren Prinzip die amerikanischen Forscher John Kasianowicz und David Deamer schon 1996 vorstellten. Die Idee der Nanoporensequenzierung ist im Grunde simpel: Eine mit feinsten Nanoporen von knapp einem Nanometer Durchmesser durchsetzte Membran trennt eine Kammer, die mit einem Elektrolyt (zum Beispiel einem Kaliumchlorid-Puffer) gefüllt ist, in zwei Kompartimente (cis und trans). Verbindet man diese mit Elektroden, so wandern die Ionen durch die Nanoporen der Membran von einer Kammer in die andere, woraus ein messbarer Stromfluss mit definierter Stärke resultiert.
Schwimmen auf einer Seite der Membran größere Moleküle, wie zum Beispiel DNA in der Lösung, so werden diese durch ihre negative Ladung ebenfalls elektrophoretisch durch die Nanoporen gezogen. Allerdings verstopfen die großen Moleküle die Poren kurzzeitig und verhindern den Ionenfluss. Diese Änderung wird gemessen und in ein auswertbares Signal umgewandelt.
Die Nanoporen bohren die Wissenschaftler durch einen Elektronenstrahl in eine künstliche Membran aus Siliziumnitrid, Graphen oder Molybdändisulfid. Man kann sich aber auch aus der Natur bedienen und porenbildende Proteinkomplexe einsetzen. Die Nanoporen-Pioniere um Kasianowicz und Deamer verwendeten alpha-Hämolysin von Staphylococcus aureus als Pore, die sie in eine Lipid-Doppelschicht-Membran integrierten. Sie wiesen nach, dass Poly A-RNA-Sequenzen andere Veränderungen des Stromflusses hervorriefen als Poly C-Sequenzen und unterschiedliche Nukleotide charakteristische Muster im Spannungsprofil verursachen.
Die beiden Amerikaner standen jedoch vor dem Problem, dass zehn bis fünfzehn Nukleotide gleichzeitig in dem gut fünf Nanometer langen Lumen der von αalpha-Hämolysin gebildeten Pore steckten und eine eindeutige Zuordnung der Nukleotide in einer DNA-Sequenz nicht möglich war. Dennoch zeichneten sich bereits die theoretischen Vorteile der Nanoporensequenzierung ab: Die Wanderung durch die Poren benötigt keine teuren Reagenzien und verläuft rasend schnell.
...aber ungenau
Es hapert aber an der Genauigkeit. Trotz zahlreicher Ansätze, wie zum Beispiel dem Einsatz von Adaptern, die Einzelnukleotide durch die Hämolysin-Pore schleusen und dadurch für A, T, C und G sehr charakteristische Signale erzeugen oder einer mit dem Hämolysin assoziierten Polymerase, die die dsDNA direkt vor den Poren in ssDNA trennt und Nukleotid für Nukleotid durch die Pore schiebt, gelang es bisher nicht, die Fehlerquote beim Sequenzieren mit αalpha-Hämolysin-Nanoporen unter vier Prozent zu drücken. Für eine verlässliche Sequenzierung ist dies nicht ausreichend.
Künstliche Festphasen-Membranen sind wesentlich dünner als alpha-Hämolysin. Mit der zweidimensionalen Kohlenstoffstruktur Graphen lassen sich zum Beispiel Membranen herstellen, die nur einen Nanometer dick sind (Garajey et al., 2013, PNAS, 110, 12092-6). Doch an Graphen bleibt die DNA kleben. Dieser Effekt tritt bei Molybdändisulfid-(MoS2)-Membranen zwar nicht auf, aber die Membran ist so dünn, dass die Nukleotide geradezu durch ihre Poren hindurch flitzen (Farimani et al., ACS Nano, 2014, 8 (8), 7914-22).
Kasianowicz äußerte sich deshalb schon 2010 gegenüber der Zeitschrift Technology Review skeptisch zu den sehr dünnen Festphasen-Membranen. Das Hauptmanko sah er in der extrem kurzen Zeit (nur wenige Nanosekunden), die die Basen für die Durchquerung der Membran benötigen.
Dieses Problem will die Gruppe von Christian Holm vom Institut für Computerphysik der Uni Stuttgart mit Computersimulationen lösen. Im Rahmen eines Sonderforschungsbereiches berechnen die Stuttgarter an Computer-Modellen, wie Pufferlösungen, Poren und Membran für die Nanoporen-Sequenzierung optimiert werden müssen (Kesselheim et al., 2014, Phys. Rev. Lett. 112, 018101).
Die Idee, Proteinkomplexe in einer Lipiddoppelschicht für die Nanoporen-Sequenzierung zu verwenden, macht angresichts der Probleme mit Festphasen-Membranen also durchaus Sinn. Die von den Proteinen geschaffenen Poren müssten nur wesentlich kürzer sein als alpha-Hämolysin.
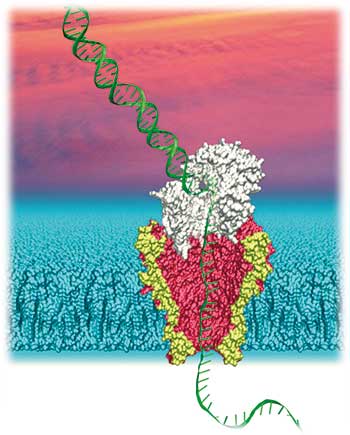
Wie man dies erreichen kann, beschrieb der Experimentalphysiker Jens Gundlach, von der University of Washington in Seattle 2012 in Nature Biotechnology (Manrao et al., Vol. 30, 349). Gundlach, der seine Karriere nach dem Physik-Diplom an der Uni Mainz 1986 in Seattle fortsetzte, favorisiert PorinA von Mykobakterium smegmatis (MspA) als porenbildendes Protein. Der MspA-Komplex hat einen Durchmesser von 1,2 Nanometer (im Lumen) und ist nur 0,6 Nanometer lang. Allerdings ist das native Protein innerhalb der Pore negativ geladen, was ein Durchwandern der ebenfalls negativ geladenen DNA verhindert.
Gundlachs Gruppe modifizierte MspA durch gezielte Mutationen der entsprechenden Aminosäuren, bis das Lumen der Pore ungeladen war und der zuführende Teil des Porenkomplexes sogar eine positive Ladung aufwies. Aufgrund ihrer idealen Größenverhältnisse ist die modifizierte MspA-Pore bestens für die Translokation von Einzelstrang-DNA (ssDNA) geeignet.
Polymerase als Schleuser
Um die Passage der DNA durch die Pore besser steuern zu können, schaltete Gundlach eine Polymerase des Bakteriophagen Phi29 vor den Porenkomplex, die den Doppelstrang direkt vor der MspA-Pore in Einzelstränge auftrennt. Die Polymerase tastet sich an einem der Stränge in 3‘- 5‘-Richtung entlang und synthetisiert einen neuen Doppelstrang. Durch die Polymerase-Aktivität, die ein Nukleotid nach dem anderen an den Matrizen-Strang anfügt, wird das 5‘-Ende, Nukleotid für Nukleotid, in die Pore geschoben. Die Gefahr, dass der DNA-Strang fragmentiert, besteht deshalb nicht.
Theoretisch ist es hierdurch möglich, sehr lange Sequenzen stabil durch die Pore zu schleusen. Das Potenzial von Gundlachs modifizierter Pore ist auch dem NGS-Giganten Illumina nicht entgangen, der im Oktober 2013 einen Lizensierungs-Deal mit der Uni Washington abschloss. In der Praxis befinden sich aber immer noch rund vier Nukleotide gleichzeitig in unmittelbarer Nähe, beziehungsweise in der Pore. Die Veränderungen des Ionenflusses durch dieses Nukleotid-Quartett erschwert eine eindeutige Zuordnung der einzelnen Nukleotide.
In Nature Biotechnology präsentiert Gundlachs Team eine elegante Möglichkeit, dies zu umgehen (Laszlo et al., Vol. 32, 829). Die Gruppe ging von folgender Überlegung aus: Wenn immer vier Nukleotide bei der Passage der Pore gleichzeitig ein elektrisches Signal auslösen, gibt es exakt 256 Kombinationsmöglichkeiten für die Reihenfolge der Nukleotide.
Buchstabenfolge löst Problem
Das Team von Gundlach synthetisierte deshalb eine 256 Nukleotide-lange DNA-Sequenz, die alle Kombinationsmöglichkeiten für diese vier Nukleotide (Quadromere) enthielt. Es stellte sich heraus, dass jedes Quadromer dieser sogenannten de Bruijn-Folge, die auch Kartenspieler für ihre Tricks benutzen, ein sehr charakteristisches und reproduzierbares Signal erzeugt. Da sich die vier Nukleotide durch das schrittweise Vorrücken der Basen in dem phi29-Polymerase-MspA-Komplex immer nur um ein Nukleotid verändern, lässt sich eindeutig sagen, welches neu zu der Quadromer-Gruppe hinzugekommen ist.
In einem Praxistest sequenzierten die Wissenschaftler das etwa fünf Kilobasen große Genom des Bakteriophagen phi X 174, dessen Sequenz bekannt ist. Die Ergebnisse speisten sie in eine Datenbank mit über 5.000 Genomen von unterschiedlichen Viren ein und fanden eine über 99,9 prozentige Übereinstimmung mit der Datenbank-Sequenz von phi X 174.
Ermutigt durch dieses Ergebnis suchte Gundlachs-Mannschaft nach Mutationen innerhalb des phi X 174-Genoms. Hierzu bauten die Forscher rund 1.000 Einzel-Nukleotid-Polymorphismen in die Sequenz ein. Immerhin 77 Prozent dieser Mutationen konnte die Gruppe mit der MspA-Nanoporen-Sequenzierung aufspüren.
Gundlach sieht für seine Membran aber noch erheblichen Verbesserungsbedarf. Bis zur fehlerfreien de-novo-Sequenzierung mit phi29-Polymerase-MspA-Nanoporenkomplexen ist es vermutlich noch ein langer Weg.
Letzte Änderungen: 04.11.2014