Mehr als nur die Summe
Röntgenstrukturanalyse, NMR und Cryo-EM
Karin Hollricher
(11.11.2020) Die Funktion biologischer Makromoleküle hängt von ihrer Struktur ab. Nur wer diese kennt, weiß, was sich
in Proteinkomplexen, molekularen Maschinen oder Ribosomen abspielt. Mit Röntgenstrukturanalyse, NMR und Cryo-Elektronenmikroskopie versuchen Biowissenschaftler, die Strukturen aufzulösen.
Strukturbiologen leben in einer spannenden Zeit. Denn heute stehen ihnen Methoden zur Verfügung, mit denen sie den Aufbau und die Funktionsweise zellulärer Moleküle in einer Geschwindigkeit und Genauigkeit untersuchen können, von der sie vor wenigen Jahren noch nicht einmal zu träumen gewagt hätten. Quasi live kann man das gerade am SARS-CoV-2-Virus mitverfolgen. In der Proteindatenbank (PDB) findet man Strukturbilder des Virus und seiner Bestandteile, die mit Röntgenkristallografie (X-Ray), Kernspinresonanzspektroskopie (NMR) und Kryo-Elektronenmikroskopie (Kryo-EM) rekonstruiert wurden.
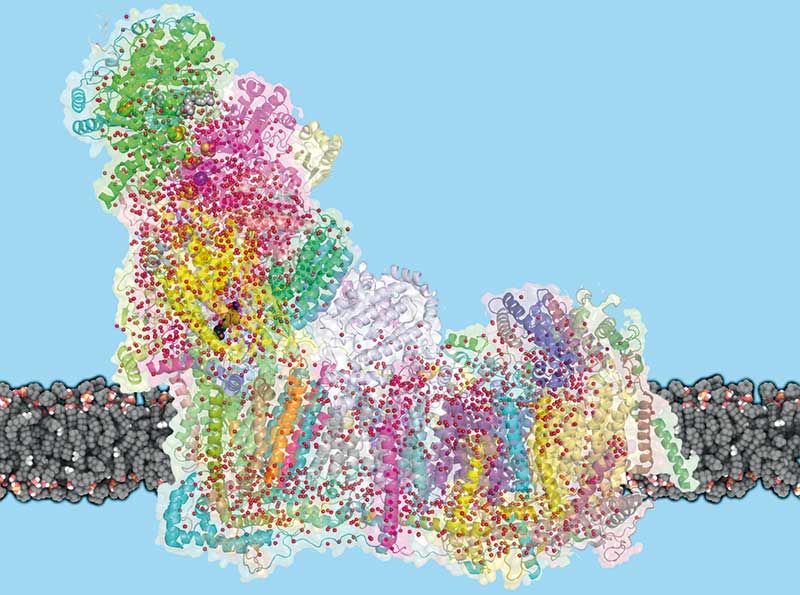
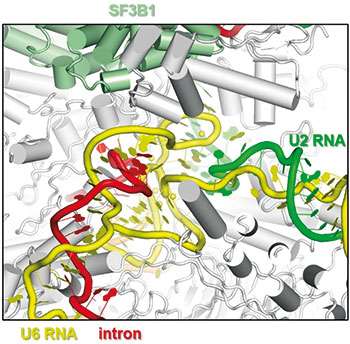
Diese drei Methoden sind die Arbeitspferde der Strukturbiologen, um Form, Größe, atomaren Aufbau und Mechanismen von Proteinen und Komplexen aufzuklären. Obwohl viele Anwender nur eine dieser Methoden favorisieren, hört man in der Strukturbiologen-Szene immer mehr Stimmen, die die Komplementarität dieser drei Techniken betonen. Ein Paradebeispiel für ihr erfolgreiches Zusammenspiel ist die Aufklärung von Struktur und Mechanismus des Spleißosoms. Dieser molekulare Komplex schneidet im Zellkern Introns aus prä-mRNA-Molekülen heraus und verbindet die verbleibenden Exons miteinander.
Forscherleben für das Spleißosom
Wie man die Struktur dieses Multi-Protein-RNA-Komplexes nach und nach enthüllte, kann Reinhard Lührmann erzählen. Er gilt als der weltweit vielleicht beste „Spleißosom-Versteher“ und verbrachte fast sein ganzes Forscherleben mit diesem Komplex. Lührmann arbeitete viele Jahre am Max-Planck-Institut für biophysikalische Chemie in Göttingen und hat dort noch immer eine Emeritus-Gruppe. „Das Spleißosom besteht aus über hundert Proteinen und fünf kleinen snRNA-Molekülen. Etwa 50 Proteine bilden mit den snRNAs Partikel namens U1- und U2-snRNP sowie U4/U6.U5-tri-snRNP. Überraschenderweise existieren Spleißosomen nicht als vorgefertigte Maschinen, sondern werden an jedem Intron aus den einzelnen Bausteinen neu zusammengesetzt, wobei sie sukzessive eine Vielzahl verschiedener struktureller und funktioneller Stadien durchlaufen. Dabei ändert sich die biochemische Zusammensetzung des Spleißosoms kontinuierlich, was eine enorme Herausforderung für die Strukturbiologie ist. Wenn man die Struktur des Spleißosoms verstehen will, reicht es nicht, einen einzigen spleißosomalen Komplex zu analysieren – man muss alle Zwischenzustände des Spleißosoms strukturell aufklären, die es während der Phasen des Assemblies, der katalytischen Aktivierung, der katalytischen Phase und des Disassemblies durchläuft.“ Dafür waren Röntgenkristallografie, NMR und Kryo-EM gemeinsam im Einsatz.
Nachdem die ersten Proteine sequenziert waren, begann man in den Neunzigerjahren mit Kristallografie und NMR, die Struktur einzelner Proteindomänen aufzuklären – später auch von größeren Proteinen sowie Protein-RNA-Komplexen. Die snRNPs stellten die Forscher allerdings auf eine harte Probe. Sie sind mit etlichen Hundert Dalton viel zu groß, um sie im NMR zu analysieren. Und bis auf eine Ausnahme, das U1snRNP, gelang es auch niemandem, sie zu kristallisieren.
Auflösungs-Revolution
Kryo-EM war damals auch nicht erfolgreich, weil man die Zwischenzustände mit prä-mRNAs in vitro assemblieren lassen musste. Daraus aber konnte man nicht genug Material für die Mikroskopie isolieren. Lührmann: „Erst in den Nullerjahren gelang es uns mit Holger Stark, die 3D-Struktur mehrerer spleißosomaler Snapshots und des tri-snRNPs bei niedriger Auflösung in den Grundzügen zu beschreiben. Mit der Entwicklung von direkten Detektoren, die die zuvor genutzten Kameras ablösten, und mit Unterstützung von sehr effizienten Maximum-Likelihood-Bildverarbeitungsprogrammen setzte dann in den ersten Jahren dieses Jahrzehnts eine Revolution in der Auflösung ein.“
Innerhalb von nur fünf Jahren wurden hochaufgelöste 3D-Strukturen aller snRNPs und der wichtigsten spleißosomalen Zwischenzustände des menschlichen und des Hefe-Spleißosoms publiziert – im Wesentlichen von drei Arbeitsgruppen in Peking, Cambridge und Göttingen.
„Damit ist ein lange gehegter Traum von mir, dem Spleißosom bei der Arbeit zuschauen zu können, erfüllt worden“, sagt Lührmann. „Während unsere früheren biochemischen Untersuchungen die kompositionelle Dynamik des Spleißosoms deutlich gemacht hatten, haben die Kryo-EM-Strukturen der diversen spleißosomalen Komplexe jetzt offenbart, wie dramatisch die Dynamik der Strukturumlagerungen des Spleißosoms beim Übergang von einem Zustand zum nächsten sein kann. So können mehrere große Strukturelemente des Spleißosoms sukzessive Translokations-Bewegungen von mehr als 20 Nanometern durchlaufen. Ein Prinzip, das von anderen molekularen Maschinen wie beispielsweise dem Ribosom nicht bekannt ist.“
Überflieger Cryo-EM
Innerhalb dieser fünf Jahre schnellte die Zahl der in der PDB archivierten 3D-EM-Strukturen von 968 auf 6.183 hoch (Stand: 23. Oktober 2020). Im Jahr 2015 wurden dort 218 Strukturen abgelegt, in diesem Jahr sind es bereits 1.899. Diese Zahlen lassen vermuten, dass Kryo-EM-Strukturforscher gerade eine ähnlich tolle Zeit haben wie Lawrence Bragg in den frühen Tagen der Röntgenkristallografie. Die beschrieb er in seiner Rede zum Nobelpreis 1950 so: „It was a wonderful time. Like discovering a new goldfield where nuggets could be picked up on the ground, with thrilling new results every week.“
Natürlich schwört auch Joachim Frank, der mit Jacques Dubochet und Richard Henderson 2017 den Nobelpreis für Kryo-EM bekam, auf die Technik: „The frontiers in structural biology are very clearly larger systems and systems in situ.“ (Biochem. 57: 277). Darum wechseln derzeit viele Forscher vom Synchrotron zum Kryo-Elektronenmikroskop.
Was wohl Max Perutz tun würde, wenn er noch lebte? Er hatte mit der Beschreibung der Struktur des Hämoglobins Anfang der Sechzigerjahre der Röntgenkristallografie den Weg in die Biologie bereitet. Seit Anfang der Achtzigerjahre liefert die NMR Daten biologischer Moleküle. Die Kryo-EM folgte kurz darauf, glänzt aber erst seit etwa zehn Jahren mit hochaufgelösten Strukturen. Deshalb stammen die meisten Strukturen in der Proteindatenbank aus der Kristallografie. Im Oktober verzeichnete man dort gut 150.000 Röntgenkristallstrukturen, rund 13.000 NMR-Modelle und etwas über 6.000 Kryo-EM-Gebilde – aber nur 177 Multi-Methods-Strukturen.
Jede der genannten Methoden hat ihre Vor- und Nachteile, ihre glühenden Verfechter und ebenso rigorosen Kritiker. Manche Forscher aber machen sich für die Kombination der Methoden stark. So will weder Lührmann in den Abgesang der Kristallografie einstimmen noch sein Kollege Holger Stark aus demselben Max-Planck-Institut – obwohl Stark gerade wissenschaftliche Erfolge mit seinem neuen Kryo-Elektronenmikroskop feiert (siehe Interview Seite 48).
Auch Oliver Einsle von der Universität Freiburg stemmt sich gegen den Niedergang der Kristallografie. „Kryo-EM ist toll, aber ihre rasante Entwicklung hat das Feld in eine deskriptive Phase zurückgeworfen“, gibt er zu bedenken. „Dabei ist sie weniger präzise als Kristallografie und enthüllt weniger chemisch-mechanistische Details als NMR. Aber ich bin zuversichtlich, dass diese Phase vorbeigehen wird und wir künftig wieder mehr an Funktion als an Struktur denken.“
Richard Henderson, der am MRC Laboratory for Molecular Biology im britischen Cambridge arbeitet, schreibt dazu in einer E-Mail: „Einzelpartikel-Kryo-EM wird in etwa vier Jahren bei den Rohkoordinaten in der Proteindatenbank die Führung übernehmen. Die Verdopplungszeit liegt bei knapp unter zwei Jahren, ist also etwas schneller als das Moore‘sche Gesetz für die Computer.“ Trotzdem glaubt er, dass die Röntgenkristallografie auf unbestimmte Zeit auf ihrem bisherigen Niveau an Synchrotrons fortgeführt werde.
NMR für die Dynamik
Auch auf die NMR wird man künftig nicht verzichten können. „Kristallografie und EM zeigen die Moleküle wie in Schnappschüssen, in denen die jeweiligen Zustände erstarrt sind. NMR aber offenbart ihre Dynamik. Zur Messung von sehr flexiblen Molekülen oder Molekülbereichen ist NMR besser geeignet als Cryo-EM. Außerdem lassen sich kurze lokale Abstände damit bestens darstellen“, sagt Gunnar Schröder vom Forschungszentrum Jülich. Er setzt auf beide Methoden, um Antworten auf seine Fragen zu finden. Seine Gruppe beschrieb erstmals die Struktur von Amyloid-ß-Fibrillen in drei Dimensionen mit hoher Auflösung. Mit ihr und mithilfe von NMR-Daten entwickelte das Team ein atomares Modell.
Am Rande der Diskussion um das Methodentrio hat sich die Kristallografie mit Freie-Elektronen-Röntgenlasern (FEL) in die Startlöcher begeben – was bisher außerhalb der Szene wenig Beachtung fand. Zwei dieser Strahlungsquellen wurden 2017 in Betrieb genommen: das European XFEL in Hamburg und das FEL am Paul-Scherrer-Institut im Schweizer Kanton Aargau. Sie können derart kurze Röntgenpulse erzeugen, dass man damit auch extrem schnelle, nur Femtosekunden dauernde Reaktionen darstellen kann. Man benötigt auch keine großen Kristalle, sondern kommt mit Mikrokristallen aus.
Alles in allem scheint es, dass die Kombination aus Röntgenstrukturanalyse, Kryo-EM und NMR mehr wert sein könnte als ihre bloße Summe. Denn jede dieser Methoden beschreibt ein Molekül aus einer anderen Perspektive, mit unterschiedlichen Details und Genauigkeit. Das sollte man sich zunutze machen.
Letzte Änderungen: 11.11.2020