Strukturen sehen statt vermuten
(01.12.2020) Holger Stark schaut sich mit der Kryo-EM die Struktur von Proteinen an. Mit einem verbesserten Mikroskop kann er sogar jedes Atom sichtbar machen.
Sie haben Biochemie studiert, sich aber auch sehr viel mit der Entwicklung und Verbesserung von Hard- und Software beschäftigt, die man für Kryo-Elektronenmikroskopie [Kryo-EM] benötigt. Wie kam das?
Holger Stark: Ich habe mit der Kryo-EM angefangen, als die Technologie dafür noch in den Kinderschuhen steckte. In Berlin, wo Ernst Ruska arbeitete, waren viele Physiker und Entwickler, die etwas von Elektronenoptik verstanden, darunter mein späterer Doktorvater Marin van Heel. Marin war eine der beiden Personen, die die Bildverarbeitungsmethoden entwickelt haben. Der andere war Joachim Frank. Beide haben parallel die ersten Arbeiten gemacht. Frank hat den Nobelpreis bekommen, aber ich finde, van Heel hätte den genauso verdient gehabt. Ich habe also Kryo-Elektronenmikroskopie und Bildverarbeitung von Grund auf gelernt. Es kam mir all die späteren Jahre extrem zugute, Biochemie studiert und eine lange Ausbildung bei Physikern genossen zu haben. Forschern fehlt es heute leider oft am Verständnis für methodische und technologische Entwicklungen, während die Techniker meistens nicht so sehr an den Anwendungen interessiert sind. Bei mir war das anders: Was ich methodisch entwickelt habe, hatte immer einen biologischen Hintergrund. Ich war zum richtigen Zeitpunkt am richtigen Ort mit den richtigen Leuten – und daher war es eine super Zeit.
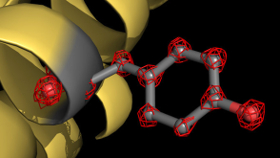
Gerade ist ein neues Paper Ihrer Arbeitsgruppe in Nature [587(7832):157-61] erschienen. Darin beschreiben Sie, dass man mit Kryo-EM jedes Atom eines Proteins sehen kann. Wie haben Sie die dafür notwendige Auflösung von 1,25 Ångström hinbekommen?
Stark: Wir haben gemeinsam mit Firmen ein besseres Mikroskop gebaut und die Bildqualität verbessert, indem wir Abbildungsfehler reduzierten. Optische Fehler entstehen nämlich in Elektronenmikroskopen genauso wie in Lichtmikroskopen als Resultat von Limitierungen und Fehlern in den Linsen. Wir haben zwei Fehler korrigiert: die sphärische Aberration und den chromatischen Fehler.
Den Proof-of-Principle haben Sie mit Apoferritin gemacht. Dieses Molekül ist sehr strukturiert, geordnet und somit exzellent geeignet für die Kryo-EM. Was ist mit ungeordneteren Molekülen mit geringerer Symmetrie?
Stark: Die Kollegen am Laboratory of Molecular Biology im britischen Cambridge haben schon vom GABA-Rezeptor eine hochaufgelöste Struktur bekommen. Wir stecken jetzt natürlich jeden Komplex, mit dem wir auch bisher schon gearbeitet haben, in unser neues Mikroskop. Aktuell haben wir Daten zu einer Fettsäuresynthase und dem Proteasom. Beide sind größer als Apoferritin und in ihrer Struktur beweglicher. Größe ist ein kritischer Punkt, weil man dann mit dickeren Eisschichten arbeiten muss, in denen die Moleküle auf dem Probenhalter eingefroren sind. Die Dicke der Eisschicht beeinflusst die Auflösung. Obendrein haben diese großen Komplexe mehr interne Flexibilität, was die Schwierigkeit erhöht, eine hochaufgelöste Struktur zu bestimmen, da nicht alle Moleküle exakt gleich aussehen.
Wie weit sind Sie dann in Sachen Auflösung gekommen?
Stark: Die beste EM-Struktur für das humane 20S-Proteasom hat bisher eine Auflösung von etwa 2,6 Ångström. Wir haben jetzt eine mit 1,8 Ångström. Für die Fettsäuresynthase haben wir die Auflösung von 2,8 auf 1,9 Ångström verbessern können. Das Spleißosom, dessen Struktur bisher auf 3,2 Ångström Auflösung abgebildet wurde, ist unser nächster Kandidat. Dafür erwarte ich auch eine deutliche Verbesserung.
Ein Ångström hin oder her, was genau bedeutet das in der Welt der biologischen Chemie?
Stark: Ich will das mal so verdeutlichen: Die Verbesserung der Auflösung von 3 auf 1 Ångström steigert die Information in den Dichtekarten um fast das Dreißigfache. Das ist gewaltig viel. Bei unter 2 Ångström beginnt eine ganz neue Welt: Man beginnt, wichtige chemische Details zu sehen statt sie nur zu vermuten. Bei 1,9 oder 1,8 Ångström sind wir noch nicht am Ende. Mit Statistik können wir die Auflösung eventuell noch auf 1,5 Ångström bringen. Allerdings ist wohl bei 1 Ångström eine Grenze, die wir mit den bisherigen Geräten nicht brechen können. Dafür müssen wir die Hardware nochmals weiter verbessern.
Wird die deutlich verbesserte Leistungsfähigkeit der Kryo-EM die Kristallografie zur Nischenmethode in der Strukturbiologie machen?
Stark: Ich hoffe nicht. Leider muss ich beobachten, dass viele Kristallografen ihr Feld verlassen und EM machen. Aber die Kristallografie ist nicht ersetzbar. Sie und EM sind komplementäre Techniken mit ihren Vor- und Nachteilen. Die Kristallografie ist immer hochauflösend und arbeitet im Hochdurchsatz. Wenn man also reproduzierbar Kristalle züchten kann, kann man mehrere Hundert solcher Kristalle täglich an einer Synchrotron-Beamline messen und die Strukturen lösen. Das geht mit Kryo-EM überhaupt nicht. Der Durchsatz ist etwa zwei Größenordnungen kleiner. Die Kristallografie aufzugeben, wäre fatal. Ich bin vielleicht einer der wenigen, der das so sagt. Es gibt viele Wissenschaftler mit starken Meinungen, die die Röntgenkristallografie schon vollständig abgeschrieben haben. Das geht mir einfach zu weit, das kann ich nicht unterstützen. Ich plädiere deshalb hier ganz entschieden nicht nur für die Verbesserung der Kryo-Mikroskope, sondern auch für den Erhalt der Synchrotrons, wenn man wirklich verstehen will, wie Proteine funktionieren und eventuell für therapeutische Zwecke inhibiert werden können.
Das Interview führte Karin Hollricher
Holger Stark studierte Biochemie in Berlin, in den Neunzigerjahren die Hochburg der Elektronenmikroskopie. Ernst Ruska, ihr Erfinder, war sein „Doktor-Großvater“. Heute ist Stark Direktor der Abteilung Strukturelle Dynamik am Max-Planck-Institut für biophysikalische Chemie in Göttingen.
Dieses hier gekürzte Interview erschien zuerst in ausführlicher Form in Laborjournal 11/2020.
Bild: Holger Stark/MPI für biophysikalische Chemie