Rettungsanker in der Daten-Flut
Microarray-Analyseprogramm Mayday
Florian Battke
Mit der Analyse von Microarray-Daten sind Informatik-Laien schnell überfordert. Eine neue Software hilft, den Überblick zu bewahren.
Wer sich schon einmal mit Microarray-Experimenten beschäftigt hat, kennt das Problem: Entweder man hantiert mit riesigen unhandlichen Tabellen oder verheddert sich in den vielen Fallstricken des für Microarray-Analysen häufig eingesetzten Statistikprogramms R. Die Arbeit mit Tabellen beschränkt sich auf das Sortieren sowie einige mathematische Operationen und die Fülle der in R implementierten Methoden macht es vielen Nicht-Bioinformatikern schwer, überhaupt ein Ergebnis zu erzielen. Der oft gehörte Notruf: "Mayday! Mayday!, wir versinken in der Flut unserer Microarray-Daten", motivierte uns dazu eine benutzerfreundliche Software zur Analyse von Microarrays zu entwickeln. Das Ergebnis unserer Arbeit ist das Programm
Mayday (Microarray Data Analysis), das von der Normalisierung der Rohdaten bis hin zu ausgefeilten Analysen viele Methoden, auch für unerfahrene Anwender, bereit hält (Battke et al.,
BMC Bioinformatics 2010, 11:121).
Gruppieren in Cluster
Eines der häufigsten Verfahren, um Gene in Gruppen mit ähnlichem Expressionsprofil einzuteilen, ist das so genannte Clustering. Man unterscheidet zwischen hierarchischen Clustering-Methoden, deren Ergebnisse eine Baumstruktur aufweisen und als Dendrogramme veranschaulicht werden, und partitionierenden Methoden, welche die Gene in nichtüberlappende Gruppen einteilen. Mayday bietet aus beiden Kategorien verschiedene Verfahren an, darunter bekanntere wie das k-Means-Clustering, aber auch weniger verbreitete, erst kürzlich entwickelte, wie das QT-Clustering. Bioinformatiker gruppieren mit diesen Clustering-Methoden nicht nur die Gene, sondern auch die untersuchten Proben, um so zum Beispiel Ähnlichkeiten zwischen verschiedenen Tumorproben heraus zu finden.
Bei Microarray-Experimenten sucht man in der Regel nach Unterschieden in Genexpressions-Mustern, zum Beispiel zwischen gesunden und pathologisch veränderten oder behandelten und nicht behandelten Zellen. Dafür gibt es in Mayday die üblichen statistischen Methoden, wie etwa den t-Test, sowie speziell für die Microarray-Analyse entwickelte Verfahren. Eines davon ist die Rangprodukt-Methode, die berücksichtigt, dass die Expression vieler Gene gemeinsam reguliert wird. Mit Maydays „Gene-Mining“-System kann man mehrere Methoden anwenden und ein Konsensus-Ergebnis berechnen. Damit läßt sich die Suche nach differentiell exprimierten Genen bei aufwendig gestalteten Microarray-Studien automatisieren.
Wir haben bei Mayday die visuelle Analyse der Daten in den Vordergrund gestellt. Im Gegensatz zu R, das eine schnelle Beurteilung und Interpretation des Analyse-Ergebnisses nur schwer zulässt, ist dieses bei Mayday sofort sichtbar. Der Anwender kann die visualisierten Daten mit wenigen Mausklicks weiter im Detail untersuchen. Alle Darstellungen (Plots) sind interaktiv und viele Eigenschaften der erzeugten Plots, etwa die farbliche Gestaltung, lassen sich anpassen, um einen klaren Überblick zu erhalten. Außerdem können zusätzliche Informationen, die sogenannten Meta-Daten, in die Darstellung einbezogen werden. Hierzu gehören Gen-Namen, Stoffwechselwege und ähnliches. Wer ein Gen genauer unter die Lupe nehmen will, kann dieses markieren, und in den verschiedenen angezeigten Darstellungen hervorheben. So erhält man schnell einen Überblick über die in einem Cluster vorhandenen Gene und kann sein „Lieblingsgen“ näher untersuchen.
Flexibles Programm
Für „Alltagsanalysen“ von Microarrays sind mit Mayday nur wenige Mausklicks nötig. Da die verschiedenen Analyseverfahren in einem einzigen Programm integriert sind, kann man neue Methoden leicht erlernen und sich die visualisierten Daten jederzeit anzeigen lassen. So erkennt man schnell Unterschiede, zum Beispiel zwischen verschiedenen Clusteringverfahren, und kann diesen weiter nachgehen. Mayday ist kostenlos und kann als Open-Source-Software nach Belieben erweitert werden. Bioinformatiker können zum Beispiel neue Analysewerkzeuge oder Software-Lösungen als Plug-In programmieren. Programmierer oder „normale“ Anwender dürfen sich gerne per Email an uns wenden, wenn sie dabei Unterstützung brauchen. Auch wir fügen neue Funktionen regelmäßig hinzu, so dass interessante neue Methoden schnell einsatzbereit sind (
www.microarray-analysis.org).
Wie man Mayday bei der Analyse von Microarray-Daten verwendet, zeigt das folgende Beispiel aus der Mikrobiologie.
Ausgangspunkt sind Microarray-Daten, die aus einer Zeitserie mit dem Boden-Bakterium Streptomyces coelicolor stammen (Nieselt et al.
BMC Genomics 2010, 11:10-10). Hierzu wurden Fermenter mit Bakterien beimpft und nach einer 20-stündigen Inkubationszeit 24 Stunden lang stündlich Proben gezogen. Anschließend entnahm man jede zweite Stunde für weitere 26 Stunden Proben. Das Nährmedium enthielt nur wenig Phosphat und nach 36 Stunden hatten die Bakterien dieses verbraucht. Dies induzierte bei
Streptomyces coelicolor das Umschalten vom primären auf den sekundären Stoffwechsel. Die damit verbundenen Veränderungen in der Genexpression wurden mit eigens für dieses Experiment entworfenen Microarrays gemessen.
So funktionierts
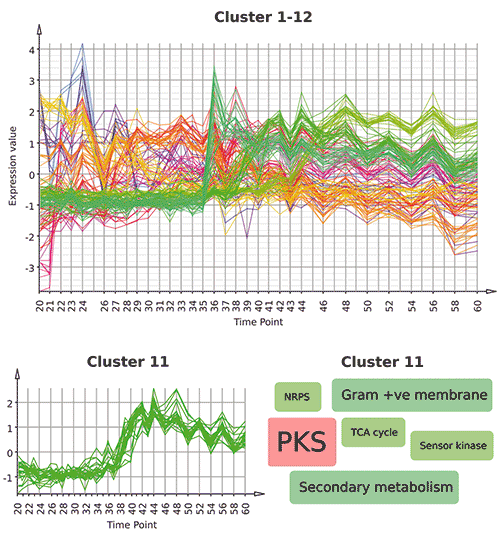
Für die Analyse der aus der Zeitserie gewonnen Microarray-Daten lädt man die Rohdaten zunächst als CEL-Dateien in Mayday und normalisiert sie mit der üblichen RMA-Methode (Robust Multichip Average). Auf einem durchschnittlich ausgestatteten Laptop dauert dies etwa zwei Minuten. Dann berechnet man die sogenannte regularisierte Varianz der Gene über den Verlauf der Zeitserie. Diese gibt an, wie stark sich die Expression eines Gens verändert, wobei man zur Normalisierung die minimale Expressionsstärke des Gens verwendet. So sind kleine Veränderungen bei stark exprimierten Genen weniger interessant als bei sehr schwach aktiven Genen. Mit einem „Filter“ wählt man Gene mit einer regularisierten Varianz von mindestens 0,1 aus und erhält 321 Gene, die man weiter analysiert. Die ausgewählten Gene gruppiert man nachfolgend mit einem entsprechenden Clusteringverfahren. In unserem Fall ist dies die relativ neue Methode des QT-Clusterings. Als Metrik verwenden wir die sogenannte Pearson-Correlation, da wir Gene mit ähnlichem Expressionsverhalten über den Verlauf der Zeitserie gruppieren wollen, unabhängig davon, wie stark diese im Durchschnitt exprimiert werden. Nach dem Clustering öffnet man mit zwei weiteren Mausklicks einen Multi-Profilplot, der die Expressionsprofile der gruppierten Gene veranschaulicht (siehe Abbildung).
Mit Mayday ist es möglich, umfangreiche Meta-Daten mit den experimentellen Daten zu verknüpfen. So kann man zum Beispiel die Sanger-Kategorien der
Streptomyces coelicolor-Gene importieren und damit jedem Gen eine Funktion zuordnen. Einen schnellen Überblick über alle verfügbaren Annotationen bietet Mayday in tabellarischer Form, wobei Gene, die man beispielsweise im Profilplot ausgewählt hat, in der Tabelle hervorgehoben werden. Spezielle grafische Darstellungen erleichtern die Interpretation zusätzlich. Erstellt man zum Beispiel eine so genannte Tag Cloud für Cluster 11, der ein interessantes Aktivierungsprofil zeigt, so sieht man, dass die häufigste Annotation der Gene in diesem Cluster „PKS“ lautet und eine weitere mit dem Namen „NRPS“ vorkommt. Dieser Cluster enthält demnach viele Polyketidsynthasen beziehungsweise nicht-ribosomale Peptidsynthetasen und spielt eine wichtige Rolle bei der Antibiotika-Produktion während des Sekundärstoffwechsels. Die genomische Position dieser Gene kann man in Maydays „Genome Browser“ untersuchen. Für die gesamte hier beschriebene Analyse sind weniger als 40 Mausklicks nötig.
Seit einigen Jahren nutzen Molekularbiologen neben Microarrays auch die Hochdurchsatz-Sequenzierung (RNAseq) für Genexpressions-Analysen. Die RNAseq hat mehrere Vorteile, unter anderem kann man mit ihr die Grenzen von Transkripten basengenau bestimmen und die Expression verschiedener Transkript-Varianten unterscheiden. Die Hochdurchsatz-Sequenzierung wird auch zunehmend für Expressionsanalysen von Spezies eingesetzt, für die keine Microarrays verfügbar sind. RNAseq-Experimente haben jedoch den Nachteil, dass sie enorme Datenmengen erzeugen und noch höhere Anforderungen an die Analyseprogramme stellen als Microarrays. Oft erschwert die schiere Menge der Daten die Auswertung mehr als sie zu erleichtern. Hinzukommt, dass bisher nur wenige benutzerfreundliche Anwendungen zur Analyse von RNAseq-Daten erhältlich sind.
Mit unserem weiterentwickelten Programm Mayday SeaSight kann man die klassischen Microarray-Fragestellungen jetzt auch mit Daten aus RNAseq-Experimenten bearbeiten. Dazu enthält SeaSight Verfahren mit denen sich RNAseq-Daten in ein Microarray-ähnliches Format überführen lassen, so dass man sie mit etablierten Analysewerkzeugen untersuchen und zusammen mit Microarray-Daten auswerten kann (Battke und Nieselt,
PLoS One 2011 Jan 31;6(1):e16345).
Letzte Änderungen: 29.04.2011