„DeepMind hat seinen Fokus auf die Verlässlichkeit der Vorhersage von Proteinstrukturen gelegt.“ - Interview Martin Steinegger zu ColaFold
Das Interview führte Henrik Müller
(12.10.2021) Martin Steinegger, Assistant Professor für Computational Biology an Seouls National University, erklärt, was AlphaFold2 in der Strukturbiologie revolutioniert und wie sich Strukturmodelle mit seiner Erweiterung ColabFold auch auf dem eigenen Laptop vorhersagen lassen.
Laborjournal: Warum ist die Strukturvorhersage von Proteinen noch immer eine Herausforderung?
Martin Steinegger » Weil ein sich faltendes Protein über eine sehr große Anzahl an Freiheitsgraden verfügt. Für kurze Polypeptide funktionieren Molekulardynamik (MD)-Simulationen zwar sehr gut. Doch unsere Formeln der klassischen Physik für Van-der-Waals-Wechselwirkungen, Wasserstoffbrücken et cetera sind alle nicht perfekt, da sie zum Beispiel keine Effekte der Quantenchemie betrachten. Auch die Energiefunktionen von Kraftfeldern, die zur Strukturvorhersage eingesetzt werden, sind nur Approximationen der Realität. Je länger ein Protein ist, umso mehr akkumulieren sich diese Ungenauigkeiten. Lange Polypeptide lassen sich deshalb nur noch ungenau vorhersagen – ganz zu schweigen von Multi-Protein-Komplexen.
Außerdem sind MD-Simulationen extrem teuer, da sie auch den Kontext sich faltender Proteine betrachten müssen, wie zum Beispiel die Energieterme des umgebenden Solvens.
Nun verwendet auch AlphaFold2 mit Primärsequenzen, Multiplen Sequenz-Alignments (MSA) und 3D-Koordinaten verwandter Proteine nichts Neues. Was ist das Revolutionäre an der Herangehensweise von DeepMind?
Steinegger » Neuartig ist sein End-zu-End-System. Vor AlphaFold2 untergliederte sich der Vorhersageprozess in einzelne Abschnitte. Eine erste Software erstellte ein MSA, die nächste berechnete aus dem MSA eine koevolutionäre Distanzmatrix, die dritte schränkte mit dieser Information den Konformationsraum des Proteins ein und so weiter. AlphaFold2 vereinigt all das in einem Schritt, wofür DeepMind tief in die Trickkiste maschinellen Lernens gegriffen hat.
Seine Neuartigkeit liegt also in bestimmten Mechanismen der Informationsweitergabe?
Steinegger » Genau. Neuronale Netze lernen, indem sie ihre finale Vorhersage – in diesem Fall ein Strukturmodell – mit einem vorgegebenen Ziel abgleichen – hier eine experimentell bestimmte Proteinstruktur. Dafür berechnen sie eine Verlustfunktion ihrer eigenen Ungenauigkeit. AlphaFold2 propagiert diese Verlustfunktion durch das gesamte neuronale Netzwerk, also vom MSA bis zum Strukturmodell, und optimiert anhand dessen seinen Vorhersage-Algorithmus. Am Ende lernt es also nicht nur, aus einem MSA 3D-Kontakte zu extrahieren, sondern, aus einem MSA 3D-Kontakte zu extrahieren, die zur Strukturvorhersage taugen. Das macht den Unterschied.
Inwieweit kann das Team von DeepMind nachvollziehen, wie die Black Box ‚neuronales Netz‘ funktioniert?
Steinegger » Als Mitautor bin ich voreingenommen. Aber ich finde, das DeepMind-Team hat einen fantastischen Job gemacht, AlphaFold2 zu verstehen. Sein neuronales Netz besteht aus zwei Blöcken, dem Evoformer gefolgt vom Strukturmodul. Der Evoformer tauscht in 48 Unterblöcken iterativ Information zwischen MSA, Distanzmatrix und Strukturvorlagen aus der Proteindatenbank (PDB) aus. Für jeden der 48 Evoformer-Blöcke schaute DeepMind mit eigens trainierten Strukturmodulen nach, was AlphaFold2 über eine Struktur hinzulernt. Abbildung 4 unserer Nature-Publikation von Juli 2021 zeigt schön, welche Blöcke wie viel Vorhersagegenauigkeit beitragen (Nature 596: 583-89).
Die 3D-Koordinaten verwandter Proteine als Vorhersagevorlage (Template) verbessern AlphaFold2s Genauigkeit nur wenig. Überrascht das nicht?
Steinegger » DeepMind verwendete die Proteindatenbank, um den Evoformer zu trainieren. Die PDB-Information ist also intrinsisch im neuronalen Netzwerk kodiert. Damit kann AlphaFold2 eine Strukturvorhersage mit großer Wahrscheinlichkeit auf eine Sequenz abbilden, die es aus der PDB kennt. Für 3D-Strukturen, die sich nicht in der PDB widerspiegeln, beeinflussen Templates aber durchaus die Strukturvorhersage.
AlphaFold2 sagt zwar Strukturen, nicht aber deren Faltungsweg voraus. Kann es trotzdem zum Wie und Warum der Proteinfaltung beitragen?
Steinegger » Für diese Fragen sind neuronale Netze zurzeit nicht der beste Ansatz, weil wir zu wenig Trainingsdaten für Faltungswege haben. Das Gleiche gilt für Fragen zur Dynamik. Letztendlich gibt AlphaFold2 die fünf besten Strukturmodelle inklusive ihrer jeweiligen Verlässlichkeit aus. Tatsächlich unterscheiden sie sich manchmal so sehr, dass sie verschiedene Proteinzustände darstellen könnten. Als Ensemble, das die Dynamik eines Proteins widerspiegelt, oder Stufen eines Faltungswegs darf man sie aber nicht interpretieren.
Mit flexiblen Sequenzbereichen tun sich Bioinformatikwerkzeuge ja generell schwer...
Steinegger » Das stimmt zwar, aber ich war trotzdem überrascht, wie hervorragend AlphaFold2 Unordnung in Proteinen voraussagt. Meine Erwartung war, dass es auch intrinsisch ungeordneten Proteinen eine Struktur aufdrückt. Schließlich hat es als Trainingsdaten nur starre Strukturen und keine komplette Unordnung gesehen. Doch die faltet es nicht.
Intrinsically disordered proteins (IDP) und unstrukturierte Domänen machen ein Drittel bis die Hälfte eukaryotischer Proteome aus. Wurde AlphaFold2 jemals mit ihnen trainiert?
Steinegger » Meines Wissens nicht. Auch hier verfügen wir nicht über genug Trainingsdaten, als dass AlphaFold2 etwas lernen könnte. Außerdem nehmen unstrukturierte Bereiche ja intrinsisch jegliche Konformation an.
Wobei IDPs ja nicht den kompletten Konformationsraum nutzen, sondern ihn einschränken. Wir verstehen nur nicht, wie. Kann AlphaFold2 helfen?
Steinegger » Ohne die Interaktionspartner von IDPs und die Natur ihrer Wechselwirkungen zu kennen, wird es AlphaFold2 schwerfallen, eine gute Verlustfunktion durchs Netzwerk zu propagieren. Zumindest scheint es schon mal vorhersagen zu können, welche Sequenz wahrscheinlich ein IDP ist und welche nicht. Das ist ja auch ein Erkenntnisgewinn.
Als Alternative zu AlphaFold2 sagt auch das neuronale Netzwerk RoseTTAFold aus dem US-amerikanischen Labor von David Baker Proteinstrukturen voraus. Worin unterscheidet es sich?
Steinegger » Bis zum CASP-Wettbewerb 2018 hatte das Baker Lab mit Rosetta eines der erfolgreichsten Vorhersageprogramme entwickelt. Doch dann schlug AlphaFold2 ja ein wie eine Bombe. Die Arbeitsgruppe um Baker begann also, AlphaFold2 nachzubauen – was ihr dank der von DeepMind veröffentlichten Details auch in extrem kurzer Zeit gelang. Beide Systeme haben daher eine ähnliche Architektur.
Was kann RoseTTAFold besser als AlphaFold2?
Steinegger » In der Publikation zu RoseTTAFold quantifizieren Baker und Co. die Strukturvorhersage von Proteinkomplexen umfangreicher. Laut eines bioRxiv-Preprints von Mitte September 2021 berechnet es Komplexe allerdings weniger gut als AlphaFold2 (bioRxiv doi: org/10.1101/2021.09.15.460468). Im Gegensatz zu DeepMinds neuronalem Netzwerk sagt es außerdem nur das Proteinrückgrat voraus, nicht aber die Seitenketten. Und es arbeitet auch mit Einzelketten noch ungenauer als AlphaFold2.
Wie haben Sie eigentlich zu AlphaFold2 beigetragen?
Steinegger » Für die herausfordernden Viren- und Phagen-Proteine der CASP-Wettbewerbe stellt die weltweit größte Proteindatenbank UniProt nur wenige Sequenzen für ein MSA zur Verfügung. Das DeepMind-Team brauchte deshalb eine metagenomische Datenbank, die die biologische Diversität besser abdeckt, da sie Millionen nicht annotierter Proteinsequenzen aus dem Boden, dem Meer, dem menschlichen Darm und so weiter enthält. Als mich DeepMind deshalb 2017 kontaktierte, hatte ich gerade 2,2 Milliarden Proteinsequenzen aus 640 Bodenproben und 775 marinen Metatranskriptomen für meinen Protein-Level-ASSembler PLASS vereint, und sie fragten mich, ob ich diese Datenbank nicht für AlphaFold2 zugänglich machen wolle.
Das klingt allein wegen der schieren Anzahl an Proteinsequenzen anspruchsvoll ...
Steinegger » Die Herausforderung bestand darin, alle Sequenzen so zu gruppieren, dass eine Suche möglichst schnell geht. Deshalb habe ich die 2,2 Milliarden Sequenzen als Multiple Sequenz-Alignments von 65 Millionen Proteinfamilien zusammengefasst, sodass eine Suchsequenz nicht mehr mit Milliarden, sondern nur mit einigen Millionen Datenbankeinträgen verglichen werden muss. Mein Beitrag zu AlphaFold2 beschränkte sich also hauptsächlich auf die Eingabedaten seines neuronalen Netzwerks in Form meiner Big Fantastic Database (BFD).
Später erweiterten Sie AlphaFold2 aber noch um eine Colab-Umgebung. Was ist das?
Steinegger » Google Colaboratory (Colab) ist eine Benutzeroberfläche für Cloud-Computing, in der sich in der Programmiersprache Python geschriebener Quellcode ausführen lässt. Dafür stellt Google Grafikprozessoren (GPU) frei zur Verfügung, über die wenige wissenschaftliche Arbeitsgruppen verfügen. Sergey Ovchinnikov, ein Science Fellow an der Harvard University, demonstrierte, wie man AlphaFold2 in Colab nur mit Primärsequenzen ohne MSAs laufen lassen kann. Eine lokale Installation entfällt damit und jeder kann AlphaFold2 auf seinem Laptop nutzen.
Also haben Sie AlphaFold2 in Colab um MSAs erweitert?
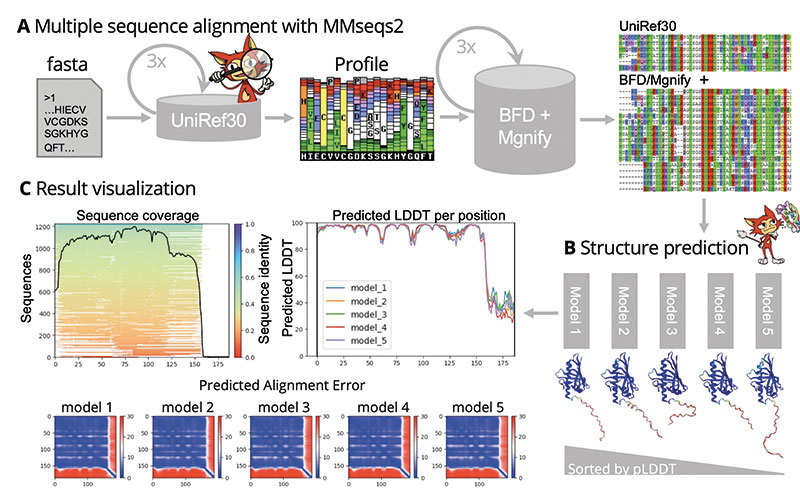
Steinegger » So ungefähr. Während meiner Promotion bei Johannes Söding am Göttinger Max-Planck-Institut für Biophysikalische Chemie hatte ich die Suchsoftware MMseqs2 für MSAs entwickelt. Zusammen mit Milot Mirdita, einem weiteren Doktoranden von Johannes Söding, setzten wir einen Server für MMseqs2 auf und verbanden ihn mit Sergey Ovchinnikovs Colab-Code. ColabFold war geboren.
Wie funktioniert ColabFold letztendlich?
Steinegger » Sie klicken auf colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipyn, geben Ihre eigene Proteinsequenz ein und klicken auf ‚Run all‘. Colab Fold schickt die Proteinsequenz daraufhin an unseren MMseqs2-Server, der durch verschiedene Datenbanken wie UniProt und BFD sucht und ein MSA zurückgibt, was dann an AlphaFold2 gesandt wird, das schließlich seine Strukturvorhersagen in Form von PDB-Dateien zurückgibt.
Erweitert haben wir das Ganze noch um Visualisierungstools – zum einen, um sich die Strukturmodelle direkt anschauen zu können, zum anderen, um die Qualität der MSAs einzuschätzen. Das ist wichtig, weil erst ein diverses MSA mit Sequenzen nah und fern der Suchsequenz die biologische Vielfalt widerspiegelt und AlphaFold2 eine zuverlässige Vorhersage erlaubt. Natürlich geben wir auch AlphaFold2s Gütemaße wieder.
Ihr bioRxiv-Manuskript zu ColabFold bescheinigt ihm für manche CASP14-Proteine sogar eine höhere Verlässlichkeit als AlphaFold2...
Steinegger » Im Durchschnitt arbeitet es nicht genauer. Manchmal reicht einfach schon eine Sequenz im MSA aus, um lDDT-, RMSD- oder TM-Werte zu verbessern. Im Vergleich zu DeepMinds Colab-Version, die durch eine reduzierte BFD sucht, ist Colab Fold aber tatsächlich verlässlicher. Seine Hauptvorteile liegen jedoch darin, auch auf weniger speicherstarken GPUs als die von DeepMind und dank MMseqs2 16-mal schneller zu laufen.
Außerdem bietet es erweiterte Optionen an. Mit ColabFold kann ein Benutzer beispielsweise AlphaFold2s Recycling-Schritte individuell erhöhen, falls Evoformer und Strukturmodul mehr als dreimal durchlaufen werden sollen. Für manche Strukturrechnungen ist das entscheidend, da sie erst nach mehreren Zyklen in ein Strukturmodell konvergieren. Darüber hinaus erlaubt es ColabFold, unterschiedliche Konsensussequenzen und Anteile eines MSA sowie eigene MSA an AlphaFold2 zu übergeben. Auch das kann die Qualität der Strukturvorhersage beeinflussen.
Was macht MMseqs2 16-mal schneller als das von AlphaFold2 verwendete Suchwerkzeug HHblits?
Steinegger » Geschwindigkeits-bestimmend in der Erstellung eines MSA ist es, alle der Suchsequenz ähnlichen Sequenzen zueinander auszurichten. In einer Datenbank wie der BFD mit Millionen Einträgen gilt es also, nicht-homologe Sequenzen schnellstmöglich herauszufiltern. Im finalen MSA landen sie sowieso nicht. Genau dafür nutzt MMseqs2 – im Gegensatz zu HHblits – einen sensitiven und schnellen Vorfilter.
Zusätzlich modelliert ColabFold auch homo- und hetero-oligomere Komplexe. Wie?
Steinegger » In den Zeilen eines MSA sind die Sequenzen des gleichen Proteins aus verschiedenen Spezies zueinander ausgerichtet. Diese Datenstruktur muss auch bei Komplexen erhalten bleiben. Im Fall eines hetero-oligomeren Komplexes sucht ColabFold deshalb nach orthologen Sequenzen aller Komplexkomponenten und fügt sie so zu einem konsekutiven MSA zusammen, dass dessen Zeilen die Sequenzen der Komplexkomponenten aus der gleichen oder von möglichst verwandten Spezies enthalten.
Im Fall eines homo-oligomeren Komplexes übergibt ColabFold mehrere identische MSAs an AlphaFold2 und gibt vor, die einzelnen MSAs wären durch eine mehrere Dutzend Aminosäurereste lange Lücke verbunden.
In beiden Fällen behandelt AlphaFold2 die Komplexkomponenten dann wie separate Polypeptidketten und kreiert den jeweiligen Komplex. Wie gut ColabFolds Vorhersagen von Proteinkomplexen sind, muss allerdings noch experimentell evaluiert werden.
Warum modelliert DeepMind nicht auch selbst Proteinkomplexe?
Steinegger » Ich glaube, weil sie zeitlich nicht alles machen können. DeepMind hat seinen Fokus auf die Verlässlichkeit der Vorhersage von Proteinstrukturen gelegt. Für ihre CASP-Teilnahme waren Protein-Protein-Interaktionen und Komplex-Modellierung entsprechend unwichtig. Interessanterweise kann AlphaFold2 Komplexe aber trotzdem modellieren, auch ohne extra dafür trainiert worden zu sein. Irgendwie erkennt es, ob eine Polypeptidsequenz Teil eines Komplexes sein kann oder nicht.
Was sind ColabFolds Schwachstellen?
Steinegger » Zwar haben wir eine Warteschleife für Dutzende Suchsequenzen implementiert, Google Colab stellt aber nur einen GPU pro Benutzer zur Verfügung. Da ColabFold entsprechend nicht parallelisiert arbeitet, empfehlen wir für Proteome einen lokalen Rechnercluster, auf dem AlphaFold2 installiert ist. Außerdem haben wir keinen Einfluss, welchen GPU ein Benutzer der Google Cloud zugeordnet bekommt. Mit einem schwachen GPU lassen sich keine langen Proteinsequenzen berechnen. Für einige US-Dollar pro Monat stellt Google Cloud aber auch speicherstarke GPUs zur Verfügung.
Apropos Cloud: Wie ist ColabFold datenschutzrechtlich abgesichert?
Steinegger » Hochgeladene Daten existieren nur im virtuellen Server der Google Cloud, auf den einzig der jeweilige Benutzer zugreifen kann. Wird eine Strukturvorhersage zurückgeliefert oder das Browserfenster geschlossen, werden auch der jeweilige Arbeitsprozess gekillt und alle Daten gelöscht. Neben der Google Cloud sehen unsere Colab Fold- und MMseqs2-Server alle Aufträge. Natürlich publizieren wir nichts davon. Inwieweit das aber Patentierungsrechte beeinflussen könnte, weiß ich nicht. Komplette Sicherheit besteht –wie bei jedem Internetdienst – nur bei lokaler Installation der 2,5 Terabyte von AlphaFold2.
Ist denn die Finanzierung der ColabFold- und MMseqs2-Server sichergestellt?
Steinegger » Da uns aktuell zwischen vier- und fünftausend Aufträge pro Tag erreichen und ColabFold seit Mitte Juli 2021 mehr als einhunderttausend Strukturmodelle berechnet hat, erscheint es uns als eine zu wichtige Ressource für die Wissenschaftsgemeinde, als dass wir sie aufgeben wollten. Gern möchten wir ColabFold deshalb als freien Service aufrechterhalten und unsere metagenomische Datenbank gleichzeitig erweitern, um sogar vertrauenswürdiger als AlphaFold2 zu werden. Eine extra Förderung erhalten wir dafür gegenwärtig nicht.