BBSome
von Mario Rembold (Laborjournal-Ausgabe 3, 2015)
Blindheit, Taubheit, Nierenschäden, fehlgebildete Hände mit zu vielen Fingern, kognitive Einschränkungen und Fettleibigkeit – dies sind einige Symptome, die im Zusammenhang mit dem Bardet-Biedl-Syndrom (BBS) auftreten können. Die Erkrankung kann unterschiedlich stark ausgeprägt sein und alle möglichen Organe betreffen. Zum Glück ist BBS sehr selten, nur eines unter 100.000 Neugeborenen ist betroffen.
Die meisten Gene, die man im Laufe der Zeit mit BBS in Zusammenhang bringen konnte, hat man durchnummeriert. Aktuell umfasst diese Liste BBS1 bis 19. Eine Mutation in nur einem davon kann das Syndrom auslösen.
Als Forscher in den frühen 2000er Jahren immer mehr BBS-Gene identifizierten, führte die Spur zu einem kleinen, aber feinen Zellorganell: dem primären Zilium. Wer nicht allzu viel mit Zellbiologie am Hut hat, denkt bei ‚Zilien’ vielleicht zunächst nur an bewegliche Wimpern, wie man sie von Pantoffeltierchen oder dem Flimmerepithel der Lunge kennt.
Unbewegliche Antennen
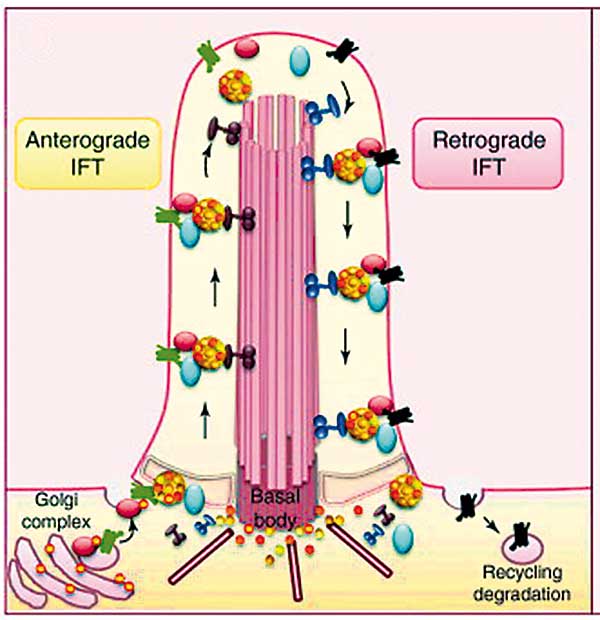
Neben diesen sekundären gibt es aber auch die primären Zilien. Diese galten bislang als nicht aktiv beweglich. Zudem fehlen ihnen die beiden zentralen Mikrotubuli, so dass sie einfach wie Antennen aus der Zelle herausragen. Und genau das ist ihre Funktion, denn sie empfangen chemische, mechanische oder optische Signale und leiten diese zur Weiterverarbeitung in die Zelle. So sitzt beispielsweise Rhodopsin in den Photorezeptoren der Retina in modifizierten Zilien. Die Haarsinneszellen im Innenohr brauchen ihre Zilien, um die Schwingungen der Basilarmembran aufnehmen und schließlich in elektrische Signale übersetzen zu können. Und während der Embryonalentwicklung konzentrieren sich Rezeptorproteine der Wnt- und Hedgehog-Signalwege in der Zilienmembran und steuern die Musterbildung. Primäre Zilien kommen in den meisten oder vielleicht sogar in allen Säugerzellen vor. Allerdings findet nach heutigem Wissensstand in den Zilien keine Proteinsynthese statt, so dass entsprechendes Material gezielt angeliefert werden muss. 2007 identifizierten Maxence Nachury et al. von der Stanford University einen Proteinkomplex, der maßgeblich an diesem Transport beteiligt ist: Das BBSome (Cell 129: 1201-13).
Kurze Zilien, fette Würmer
Das BBSome ist an der Basis primärer Zilien lokalisiert und enthält sieben der bereits zuvor charakterisierten BBS-Proteine und ein damals neu entdecktes Protein namens BBIP10. Der Proteinkomplex ist hoch konserviert und kommt in allen Organismen mit Zilien vor – von der Grünalge Chlamydomonas bis hin zum Menschen. In Pilzen, Amöben und Pflanzen, also den Eukaryoten ohne Zilien, findet man hingegen kein BBSome. Stört man die Funktion des BBSome-Komplexes durch Knockout, zeigt selbst der Fadenwurm Caenorhabditis elegans einen Phänotyp, der an BBS-Patienten erinnert: Die Würmer werden fettleibig, und ihre Zilien sind deutlich kürzer als beim Wildtyp. Die Biogenese der Organellen ist gestört, weil der intraflagellare Transport (IFT) von Proteinen nicht mehr korrekt abläuft. Am IFT sind Proteine beteiligt, die das zentrale Gerüst aus den neun Mikrotubulipaaren aufbauen und erhalten. Das BBSome ist darüber hinaus aber auch notwendig, um Membranproteine in die Zilien zu befördern und damit die Sensibilität dieser zellulären Antennen für ein bestimmtes Signal herzustellen.
Ein kleiner Helfer
Proteine, die für die Zilienmembran bestimmt sind, stecken zunächst in Membranen von Vesikeln, die sich vom Golgi-Apparat abschnüren. Diese Vesikel wandern zur Basis des Ziliums und verschmelzen dort mit der Membran. Bereits 2001, sechs Jahre vor Entdeckung des BBSomes, zeigten US-Forscher am Krallenfrosch, dass für die Fusion dieser Vesikel mit der Membran das kleine G-Protein Rab8 in seiner aktivierten Form notwendig ist. Gibt es in der Zelle nur die künstlich eingebrachte, konstitutiv inaktive Variante, dann gelangt Rhodopsin nicht in die Zilien und es kommt zu einer Degeneration der Retina (Mol Biol Cell 12: 2341-51).
Inaktives Rab8 schwimmt im Cytoplasma und ist mit einem GDP-Molekül versehen. Zur Aktivierung benötigt Rab8 ein Protein, das sein GDP gegen ein GTP austauscht. Ein solcher Austauschfaktor ist Rabin8. Rabin8 bindet an das BBSome und steht dann an der Basis des Ziliums zur Verfügung. So kann Rab8 über das BBSome aktiviert werden, und die wertvolle Fracht gelangt in die Zilienmembran.
Wanderkomplex
Jüngst schaute sich das Team um Esben Lorentzen am Martinsrieder MPI für Biochemie die Interaktion mit Arl6 an, einem anderen kleinen G-Protein. Arl6 bindet an das BBSome und hält dieses in der Nähe der Zellmembran. Allerdings kann nur die GTP-Variante von Arl6 das BBSome rekrutieren. Lorentzen und seine Kollegen haben Kristallstrukturen der Proteinkomplexe analysiert und festgestellt, dass bestimmte Mutationen im BBS1-Gen des BBSomes die Bindung zu Arl6 verhindern, selbst wenn dieses mit einem GTP versehen und damit aktiviert ist. Und eben jene Mutationen findet man in 30 Prozent aller BBS-Patienten. (Nat Struct Mol Biol 21: 1035-41). Übrigens ist auch Arl6 ein alter Bekannter: das Gen zu diesem Protein ist unter dem Namen BBS3 dokumentiert und konnte bereits 2004 mit dem Bardet-Biedl-Syndrom in Zusammenhang gebracht werden.
Schließlich sei noch erwähnt, dass das BBSome nicht ausschließlich an der Basis von Zilien lokalisiert ist, sondern auch in die Zilien eindringt, dort bis zur Spitze und wieder zurück wandert – und dabei den intraflagellaren Transport unterstützt (Nat Cell Biol 14: 950-7). Auch für das BBSome gilt also: Klein, aber agil.
Letzte Änderungen: 12.03.2015