Virtueller Talkmaster
Interpretation von Sequenzvarianten mit GeneTalk
Alexej Knaus
Bei der Annotation von Sequenzvarianten ist die Expertise von Forschern gefragt. Über GeneTalk können sie sich weltweit austauschen.
Überall liest man, dass die Next-Generation Sequenzierung auf dem Vormarsch ist und gesamte Genome von Patienten sequenziert werden. Doch was nützt es, wenn man das genetische Buch eines Patienten mit seinen drei Milliarden Buchstaben liest aber nicht versteht?
Die Herausforderung liegt darin, die wesentlichen Textpassagen des Buches zu entschlüsseln. Dabei konzentriert man sich auf die Abweichungen von der Textvorlage, das heißt auf die individuellen Besonderheiten eines Humangenoms. Die Referenz-Sequenz, derzeit hg19, ist so etwas wie der größte gemeinsame Nenner aller menschlichen Genome, die Abweichungen im Genom eines Patienten zu dieser Sequenz sind seine persönliche Signatur. Betrachtet man nur die proteinkodierenden Abschnitte, das Exom, so findet man immerhin 20.000 bis 30.000 Sequenzvarianten. Will man die molekulare Ursache einer monogenetischen Erkrankung identifizieren, muss man die Genveränderungen finden, die dafür verantwortlich sind.
Hierzu sequenziert man zunächst eine Probe des Patienten, was zu einer Flut von Daten führt: die reinen Sequenzrohdaten, üblicherweise im FastQ-Format, das Sequenzalignment, meist im BAM-Format und die Sequenzvarianten, idealerweise im Variant Call-Format, VCF.
Die Nadel im Heuhaufen
In der VCF-Datei sind alle Abweichungen von der Referenz geordnet nach Chromosom und Position aufgelistet. Man könnte die Datei in Excel öffnen und für jede Variante prüfen, in welchem Gen sich die Abweichung befindet, ob sie die Proteinsequenz verändert und ob in der Literatur schon etwas dazu zu finden ist. Bei rund 25.000 Varianten in einem Exom würde das jedoch zu lange dauern. Auf der Suche nach krankheitsverursachenden Veränderungen benötigen Lebenswissenschaftler und Ärzte die Hilfe eines Bioinformatikers oder einer Software, die diese Information automatisch hinzufügt. Die Interpretation der Daten verlangt jedoch Expertenwissen, das man sich für jedes Gen und jede Variante in mühevoller Kleinarbeit aneignen müsste.
Um dies zu erleichtern, haben wir am Institut für Genetik der Charité in Berlin die Software „GeneTalk“ für Genetiker und andere Biowissenschaftler entwickelt (www.gene-talk.de). Den Ursprung hat GeneTalk in wöchentlichen Treffen am Institut für Genetik, bei denen Kliniker und Wissenschaftler zum Beispiel die molekulargenetischen Befunde nach einer Exomsequenzierung diskutierten. In den meisten Fällen saß ein Experte in der Runde, der erklären konnte, warum ausgerechnet diese oder jene Variante die vielversprechendste war. Dieses Experten-Knowhow wollten wir auf einer Onlineplattform bereitstellen (Kamphans, et al., Bioinformatics, 28, 2515-6).
GeneTalk ist eine webbasierte Plattform für Genetiker, die das Filtern und Priorisieren, sowie die Interpretation von NGS-Daten vereinfacht. Ausgeklügelte Algorithmen erlauben den GeneTalk-Nutzern auch Datensätze von mehreren Familienmitgliedern hinsichtlich verschiedener Vererbungsmodelle zu filtern (Kamphans et al., PloSOne 8, e70151) und ihre Qualität zu beurteilen (Heinrich et al., Genome Medicine 5, 69). Darüber hinaus erleichtern eingebundene, öffentlich zugängliche Datenbanken (etwa dbSNP, 1kGP, ESP6500) die Daten-Interpretation.
Versammeltes Expertenwissen
Die wahre Stärke von GeneTalk sind jedoch die Annotationen der weltweiten Nutzergemeinde. Jeder Nutzer kann Kommentare und Bewertungen für unbekannte Varianten hinterlassen und den Abschnitt im Genom markieren, zu dem er als Experte kontaktiert werden kann.
In der Diagnostik von heterogenen, genetischen Erkrankungen findet derzeit ein Umdenken statt – weg von aufeinanderfolgenden Einzelgenuntersuchungen hin zu Exom- und Genomsequenzierungen. Bei prädiktiven oder vorgeburtlichen genetischen Untersuchungen werden NGS-Verfahren derzeit selten eingesetzt. Der Hauptgrund hierfür ist die unvollständige Interpretierbarkeit der anfallenden Zufallsbefunde.
Bislang werden genetische Tests auf schwere Erkrankungen wie familiärer Brustkrebs, Chorea Huntington oder Tay-Sachs-Syndrom (TSD) nur Personen angeboten, die ein erhöhtes Risiko tragen. Bei Paaren mit Kinderwunsch sind TSD-Tests ohne erhöhte Prädisposition jedoch medizinisch nicht indiziert. Ähnlich ist es bei prädiktiven Tests für Brustkrebs und Chorea Huntington.
Die Exomsequenzierung ist mit einem multiplen Testverfahren vergleichbar, das auf einen Schlag viele bekannte, pathogene Mutationen aufspürt, die schwere Erkrankungen hervorrufen können. Damit steigt die Wahrscheinlichkeit zumindest in einem Gen eine auffällige Veränderung zu finden. Im Unterschied zum Test einzelner Gene, bei dem fast nur bei genetischer Prädisposition auffällige Ergebnisse zu erwarten sind, weisen Exom-Untersuchungen bei den meisten Personen Befunde auf, die in einer genetischen Beratung besprochen werden sollten.
Belanglos oder Gesundheitsrisiko?
Derzeit finden sich in jedem neu sequenzierten Exom rund 300 unbekannte Varianten (Tennessen et al., Science 337, 64-9). Selbst wenn alle Menschen bereits untersucht wären, kämen mit jedem neuen Individuum bis zu 60 Neumutationen pro Genom hinzu (Roach et al., Science 328, 636-9). Die meisten dieser Sequenzvarianten haben jedoch keine schwerwiegenden gesundheitlichen Folgen. Über GeneTalk können Experten die medizinische Bedeutung dieser Varianten diskutieren und bewerten.
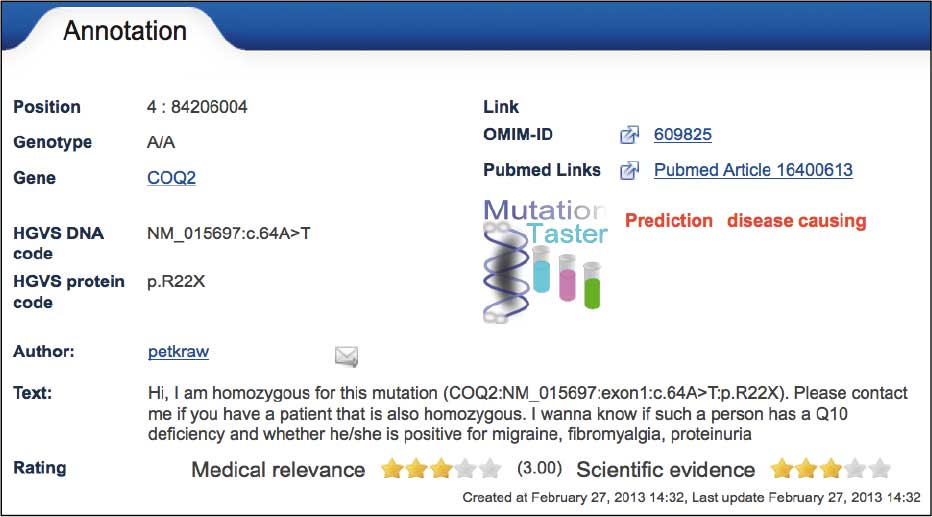
Im Personal Exome Project (PEP) von GeneTalk untersuchen Humangenetiker ihre eigenen Exome. Die ersten hieraus gewonnenen Erfahrungen verdeutlichen die Grenzen der aktuellen Vorhersagemöglichkeiten. Peter Krawitz von der Computational Biology Group der Charité trägt zum Beispiel eine homozygote Stopp-Mutation im Gen COQ2: (http://gene-talk.de/annotations/776520). Dies müsste theoretisch zu einer unvollständigen Synthese des kodierten Proteins und zu einem schweren Coenzym Q10-Mangel führen. Die üblichen Vorhersagetools würden diese Mutation deshalb als pathogen einstufen. Peter ließ darauf hin seine Coenzym Q10 Werte bestimmen. Da sie nachweisbar waren und er bislang über keine der für Coenzym Q10-Mangel beschriebenen, schweren Symptome klagt, muss man die Einstufung als Pathogen kritisch hinterfragen.
Schwierige Prognosen
Malte Spielmann, ein weiterer Teilnehmer des PEP, entdeckte in seinem Exom eine Variante im Gen SCARF2, die das Van-Den-Ende-Gupta-Syndrom auslösen sollte (http://gene-talk.de/annotations/886423). In einer genetischen Beratung würde dies zu einem Dilemma führen. Einerseits sollten Ratsuchende über den Befund und seine möglichen Auswirkungen aufgeklärt werden – wenn sie es wünschen. Andererseits können Genetiker die Auswirkungen auf die Gesundheit bei vielen seltenen Varianten noch nicht einschätzen. In diesem Fall konnte durch einen weiteren Teilnehmer des PEP, Alexej Knaus, zumindest teilweise Entwarnung geben werden. Auch er trägt diese Gen-Variante und ist dennoch symptomfrei. Da das Syndrom sehr selten ist, sinkt damit die Wahrscheinlichkeit, dass es sich um eine pathogene Mutation handelt.
Gerade Forschungsergebnisse, die eine wissenschaftliche These nicht unterstützen, erscheinen selten in den klassischen, medizinischen Publikationsmedien. Für die genetische Diagnostik sind sie jedoch von sehr großem Wert. GeneTalk kann als interdisziplinäre Plattform helfen, Forschung und Klinik näher zusammen zu bringen, um genetische Veränderungen, die mit den neuen Sequenziertechnologien in großen Mengen detektiert werden, besser interpretieren zu können.
Letzte Änderungen: 30.08.2013